
Abstract
To guarantee food safety, a better deciphering of ecology and adaptation strategies of bacterial pathogens such as Salmonella in food environments is crucial. The role of food processing conditions such as cleaning and disinfection procedures on antimicrobial resistance emergence should especially be investigated. In this work, the prevalence and antimicrobial resistance of Salmonella and the microbial ecology of associated surfaces communities were investigated in a pig slaughterhouse before and after cleaning and disinfection procedures. Salmonella were detected in 67% of samples and isolates characterization revealed the presence of 15 PFGE-patterns belonging to five serotypes: S.4,5,12:i:-, Rissen, Typhimurium, Infantis and Derby. Resistance to ampicillin, sulfamethoxazole, tetracycline and/or chloramphenicol was detected depending on serotypes. 16S rRNA-based bacterial diversity analyses showed that Salmonella surface associated communities were highly dominated by the Moraxellaceae family with a clear site-specific composition suggesting a persistent colonization of the pig slaughterhouse. Cleaning and disinfection procedures did not lead to a modification of Salmonella susceptibility to antimicrobials in this short-term study but they tended to significantly reduce bacterial diversity and favored some genera such as Rothia and Psychrobacter. Such data participate to the construction of a comprehensive view of Salmonella ecology and antimicrobial resistance emergence in food environments in relation with cleaning and disinfection procedures.
Similar content being viewed by others

Introduction
Salmonellosis remains one of the main foodborne zoonosis in Europe with 94.530 cases in 20161. In France, between 2008 and 2013, Salmonella was the leading cause of death related to contaminated food (26.2%)2. Pig products constitute common sources of human infections involving Salmonella and slaughtering line represent a critical stage for controlling its dissemination on the food chain3,4. The continuous introduction of bacteria, potentially including Salmonella, with each new pig band and the presence of a resident flora associated to the slaughter environment may indeed constitute an important risk for carcass contamination during the slaughtering or meat-cutting processes5 despite good hygiene practices. In order to guarantee food safety, a better understanding of Salmonella ecology and persistence strategies in such food environment constitutes a prerequisite. In this perspective, the role of interactions with resident flora in pathogenic Salmonella survival on the food chain6 require a particular interest. Indeed, several studies have showed that formation of biofilm and persistence of Salmonella could be promoted by the presence of other bacteria including strains directly isolated from food industries7,8. However, data about microbial ecology in pig slaughterhouses are lacking in the literature, thus stressing the need to improve our knowledge on microbial diversity and population dynamics in such food environments.
Alongside, antimicrobial resistance (AMR) in bacteria including foodborne pathogen as Salmonella is becoming one of the most preoccupying issues for public health. Being a continuum between environment, animals and human health, food chain constitutes a privileged area for occurrence and spreading of antimicrobial resistance9,10. Food processing industries as slaughterhouses for instance can act as connecting paths including substantial selection for resistance due to procedures inherent to the industrial process such as cleaning and disinfection (C&D). In the last few years, an increasing number of studies have indeed highlighted the potential role of biocides in the selection of cross-resistance towards antibiotics in bacteria11,12,13. Nevertheless, the vast majority of them were conducted using pure culture models in laboratory conditions, and the current lack of data makes it difficult to evaluate the substantial risk to select bacteria with higher resistance to antibiotics through the use of biocides in industries and to determine which biocides is associated with the highest risk of antibiotic resistance cross-selection. The need to multiply environmental studies focusing on the characterization of cross-resistance to antibiotics in pathogens following use of biocides and including considerations about complex microbial ecology associated was therefore highlighted14.
In this context, this project aimed at collecting data about Salmonella prevalence and anti-microbial resistance (AMR) levels in a pig slaughterhouse, gaining an overview of associated bacterial community diversity and to examine how C&D steps impact microbial communities and potentially select isolates with reduced susceptibility to biocides and/or antibiotics.
Materials and Methods
Pig slaughterhouse sampling and samples processing
The study was carrying out in a French pig slaughterhouse. The slaughterhouse was visited four times between March and May 2017. Samples were collected from the 6 following areas: 2 were located from the “dirty” zone at the dehairing machine (DH) and whips from the scraping machine (WH), and 4 were taken from the “clean” zone (after a singeing step) on the neck clipper machine (NC), carcass opener circular saw (CO), white offals gutter (WOG) and platform used for red offal removing (ROP). Each area was sampled twice at the same location for each sampling date, before and after C&D procedures. While the cleaning and disinfection procedures are highly dependent from the slaughterhouse, the pig slaughterhouse sampled in this work was chosen because it used biocide formualtions among the most used. Concretely, C&D procedures consisted here in daily application of a chlorinated alkaline solution along chain using foamer after slaughtering following by a rinsing step. An ethanol-based solution was additionally sprayed on cutting blades in contact with meat at NC and CO areas. In addition, an acid foaming solution was applied weekly along the slaughter chain. Samples were collected by swabbing 1 m² of surfaces using sterile wipes and placed individually in sterile Stomacher bags with filters (BagFilter® P400, Interscience, France) which were then transported to the laboratory in refrigerated conditions and processed within 12 h. Upon arrival, bags containing wipes were filled using 225 ml of One broth medium (Oxoid, France) and introduced in a stomacher (BagMixer® 400 P, Interscience, France) for blending during 1 min at high speed. Two aliquots of 20 ml of stomached suspension were directly sampled from bags and centrifuged at 10,000 g during 5 min and pellets were frozen at −80 °C for further DNA extraction and 16s rRNA sequencing. An additional aliquot of 1 ml was used to enumerate total bacterial flora after serial 10-fold dilutions in peptone water and plating on tryptone soya agar (TSAye, Beckton-dickinson, France). Differences in bacterial counts between sampling area or before and after disinfection were analyzed statistically using analysis of variance (ANOVA, significance for p < 0.05). One aliquot of 10 ml was also directly frozen at −80 °C for sample conservation.
Salmonella enrichment, isolation and PCR confirmation
The Salmonella PrecisTM protocol validated by AFNOR to ISO 16140 standard was used according to manufacturer instructions for the enrichment, detection and confirmation of Salmonella. Concretely, after stomaching, bags containing wipes in One broth medium (Oxoid, France) were transferred at 42 °C for enrichment. After 20–24 h, 10 µl were plated on Brilliance Salmonella agar (Oxoid, France) and incubated 24 h at 37 °C before identification of positive Salmonella colonies according to manufacturer instructions. Salmonella were then re-isolated on TSAye and incubated 20 h at 37 °C before being stocked at −80 °C in cryoprotective solution or used for PCR confirmation. PCR was performed using forward primer ST11 5′-GCCAACCATTGCTAAATTGGCGCA-3′ and reverse primer ST15 5′-GGTAGAAATTCCCAGCGGGTACTGG-3′15 and a LightCycler 480 thermocycler (Roche, France) as follows: 95 °C for 5 min for initial melting; 40 cycles at 95 °C for 30 s, 59 °C for 30 s, 72 °C for 30 s; and 72 °C for 10 min following by incubation at 4 °C.
Serotyping
Serotyping was performed according to the White-Kauffmann-Le Minor scheme (refs. 16,17).
Pulse field gel electrophoresis (PFGE)
PFGE using XbaI restriction enzyme was carried out with a CHEF-DR III system (Bio-Rad), according to the PulseNet Protocol18. Salmonella enterica serotype Braenderup H9812 was used as the molecular size marker in the PFGE experiment19. Gels were stained with ethidium bromide and banding patterns were visualized under UV light, using the Gel Doc XR and Quantity One software (Bio-Rad). DNA patterns were analyzed with BioNumerics software (V 7.6.3, Applied Maths, Kortrijk, Belgium). Algorithms available within the program were used to compare patterns. Dendrograms were produced, using the Dice coefficient and the unweighted pair group method with arithmetic averages (UPGMA), with a 1% tolerance limit and 1% optimization20. Each PFGE pattern differing by at least one band from a previously recognized type was considered to be a new pattern. Each new pattern was given a unique designation, as suggested by Peters et al.21, and added to the PFGE patterns library. The recommendations of Barrett et al.22 were also followed for the interpretation of PFGE patterns.
Biocide susceptibility testing
Minimal inhibitory concentrations (MIC) were determined using a standard microdilution method for the 3 biocide solutions used in the slaughterhouse: a chlorinated alkaline solution, an ethanol-based solution and an acid foaming solution. Bacterial inocula were prepared from overnight cultures diluted in Muller-Hinton broth to reach from 1 to 3.105 CFU/well in presence of various concentrations of biocides in a 96-wells microtiter plate (Greiner Bio-one 650101, France). Plates were then incubated at 37 °C for 24 h and MIC was determined as the lowest concentration of biocide that prevents bacterial growth. All determinations of MIC were repeated twice. Statistical differences between MIC distribution for biocides between strains isolated before and after C&D procedures or between serotypes were assessed using a Student test (significance for p < 0.05).
Antibiotic susceptibility testing
Antibiotic susceptibility tests were performed using a standard microdilution method with the Sensititre® system on EUVSEC plates (Trek Diagnostic Systems, UK) using a panel of 14 antimicrobial substances according to manufacturer instructions. The strains were interpreted as resistant to antibiotics according to the epidemiological resistance cut-off determined from EUCAST (European Committee on Antimicrobial Susceptibility Testing, http://mic.eucast.org).
DNA extraction, PCR amplification and 16s rRNA sequencing
Frozen pellet obtained from 20 ml sample extract after stomaching were centrifuged and DNA extraction was performed on the pellet. Nucleospin® tissue kit (Macherey-Nagel) was used for the extraction according to manufacturer instructions. Concentration and quality of extracted DNA were checked using a BioSpec-nano spectrophotometer (Shimadzu). The bacterial V3-V4 region of the 16S rRNA gene was then amplified using the forward primer 5′-CTTTCCCTACACGACGCTCTTCCGATCTACGGRAGGCAGCAG-3′ and the reverse primer 5′-GGAGTTCAGACGTGTGCTCTTCCGATCTTACCAGGGTATCTAATCCT-3′23. The preparation of amplicons was performed in a total volume of 50 µL containing 0.5 µl of TAQ Polymerase (5 U/µl) and 5 µl of adequate 10 X PCR buffer (MTP Taq DNA Polymerase, Sigma), 1 µl of 10 mM dNTP (Sigma), 1.25 µM of each primer (20 µM) and 10 ng of DNA template. PCR was performed using a LightCycler 480 thermocycler as follows: 94 °C for 60 s for initial melting; 30 cycles at 94°c for 60 s, 65 °C for 60 s, 72 °C for 60 s; and 72 °C for 10 min following by incubation at 4 °C. Products were then verified on a 1.5% agarose gel before being sent to GeT-PLaGe Genotoul plateform (Castanet-Tolosan, France) for paired-end sequencing on Illumina MiSeq platform (Illumina, USA) at a read length of 2 × 250 pb.
Bioinformatics analyses
Illumina sequences were processed using the FROGS pipeline (Find Rapidly OTU with Galaxy Solution)24 implemented on a galaxy instance (http://migale.jouy.inra.fr/galaxy/). Bacterial 16S rRNA paired-end reads were merged with a maximum of 10% mismatches in the overlap region using FLASH25. Denoising procedures consisted in discarding reads with no expected length and the ones containing ambiguous bases (N). After dereplication, the clusterisation tool ran with SWARM26 with an aggregation distance equal to 3. Chimeras were then removed using VSEARCH27 and sequences were filtered to keep OTUs (also called clusters) accounting for at least 0.005% of all sequences28. Taxonomic affiliation was performed with both RDP Classifier29 and Blastn+30 against the SILVA 132 pintail 100 database31.
Data set was rarefied and alpha diversity indexes (observed OTU, Chao1, Shannon and InvSimpson) were calculated and further plotted using the R “phyloseq” package32. Principal coordinate analysis (PCoA) were performed on weighted unifrac dissimilarity matrices calculated using the R package phyloseq. Using non-parametric permutation-based multivariate analysis of variance (PerMANOVA, function adonis in R package “vegan”) on unifrac and weighted unifrac-based distance matrices, we tested for significant differences in community structure. Assumptions of the adonis test were verified using the betadisper function in the R package vegan, which tests the multivariate homogeneity of group dispersions. Pairwise comparisons were performed on weighted unifrac distance matrix using pairwise.adonis function from “vegan” package and Bonferroni p-value correction (significance for p < 0.05). Figures were prepared with R and the package ggplot2.
Differentially abundant taxa in bacterial populations depending on various factors were identified using the linear discriminant analysis (LDA) effect size (LEfSe)33. Relative abundances of all features were first compared by using the nonparametric Kruskal-Wallis rank sum test (significance for p < 0.05), and each statistically significant feature was further subjected to effect size estimation using LDA.
Results
Sampling, isolation and typing of Salmonella
A total of 48 wipes taken before (24) and after (24) disinfection at six different sites in a pig slaughterhouse and at four different dates were collected. Total flora enumeration showed similar bacterial counts for the different areas sampled with values comprised between 3.34 ± 1,87 and 4.68 ± 0.56 log (CFU)/cm² except for the dehairing area (DH) before C&D procedures which exhibited significant higher bacterial counts with 6.15 ± 0.26 log (CFU)/cm² (see Table 1). There was no significant difference between bacterial counts between samples taken before and after C&D, except for the dehairing area with a significant decrease (p < 0.05) from 6.15 ± 0.26 to 4.66 ± 0.79 log (CFU)/cm² after C&D.
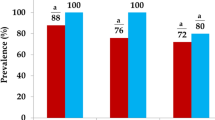
A total of 38 Salmonella strains were isolated from 32 samples (67% of positive samples) with 18 and 20 strains isolated before and after C&D procedures respectively. There was no significant difference between numbers of positive samples before and after C&D procedures. All sites investigated were positive for Salmonella except the neck clipper for which Salmonella was never isolated (Table 1). PFGE and serotyping analyses revealed 15 PFGE patterns belonging to the following 5 serotypes: S. 4,5,12:i:-, Typhimurium, Rissen, Infantis and Derby. The monophasic Typhimurium variant 4,5,12:i:- represented 58% of strains isolated and showed 9 distinct PFGE patterns (B01, B02, B03, B04, B05, B06, B09, B15 and B16), serotype Typhimurium represented 13% of isolates and showed 1 PFGE pattern (B09), serotype Rissen accounted for 10.5% of isolated Salmonella and presented 2 PFGE patterns (B10 and B11), serotype Infantis reached 10.5% of the total number of isolates with a unique PFGE patterns (B12) and serotype Derby represented 8% of strains isolated and was subdivided in 3 PFGE patterns (B08, B13 and B14). As showed in Table 1, while monophasic Typhimurium variant serotype 4,5,12:i:- was found at all Salmonella-positive sampling area and for various dates, serotype Typhimurium was only isolated at a single date (04/24) at DH, WOG and ROP areas. Interestingly, serotype Infantis was exclusively found at CO area but repeatedly for three of the four dates investigated. Serotypes Rissen and Derby were occasionally isolated both at two different dates (03/22 and 04/03) and sampling areas (WH and WOG for Derby and WOG and ROP areas for Rissen).
Antibiotic resistance profiles
Antibiotic susceptibilities of the 38 isolated Salmonella strains were obtained for a total of 14 antibiotics. MIC values obtained are available in Table 2. Resistances to ampicillin, sulfamethoxazole and tetracycline were observed for all PFGE patterns of the monophasic Typhimurium variant 4,5,12:i:- serotype except for the PFGE pattern B02 which was susceptible to all antibiotics tested. Two of the three PFGE patterns of the Derby serotype exhibited resistance to sulfamethoxazole and tetracycline, the third being susceptible to the 14 antibiotics (type B13). Strains of serotype Rissen were all resistant to tetracycline. Strains of serotype Typhimurium were resistant to ampicillin, chloramphenicol, sulfamethoxazole and tetracycline. Strains from serotype Infantis were susceptible to all tested antibiotics. There was no marked increase in number of resistant strains comparing isolates before and after C&D procedures for the different antibiotics tested. 66.7 and 70.6% of isolated strains were resistant to ampicillin, 16.7 and 11.8% to chloramphenicol, 77.8 and 70.6 to sulfamethoxazole, 83.3 and 82.4 to tetracycline before and after C&D, respectively. In addition, none of the persistent PFGE patterns which were isolated repeatedly at a given area both before and after C&D showed a significant change in MIC values of more than one dilution for the 14 antibiotics tested (Table 2).
Susceptibility of Salmonella isolates to biocides
MICs of the three biocide solutions used in the slaughterhouse were determined using a broth microdilution method. MIC distributions for the Salmonella isolated before and after C&D procedures are displayed in Fig. 1 for the three biocides used in the slaughterhouse. MIC values were comprised between 0.1 and 1.6% for chlorinated alkaline solution, 0.05 and 0.4% for acid solution and 12.5 and 50% for ethanol-based solution. There was no significant difference (p > 0.05) when comparing MIC distributions between strains isolated before and after C&D for these three biocides. C&D procedures used here thus did not lead to the selection of strains with lower susceptibility toward biocides during the two months of observation. There was also no significant difference between the serotypes for the different biocide tested (p > 0.05).

Minimal inhibitory concentrations (MIC) for the three biocides used in the slaughterhouse. Distribution of MIC values for the chlorinated alkaline solution, acid solution and ethanol base solution are compared before and after C&D.
Microbial surface community diversity
A total of 47 samples were sequenced successfully (the sample from the 03/22 before C&D at the carcass opener area (CO) did not yield sufficient DNA for sequencing). After filtering for quality, length and removal of chimera, a total of 1 175,969 sequences were obtained corresponding to an average read number per sample of 25,020 ± 11,073 reads. A total of 441 operational taxonomic units (OTUs) were obtained after bioinformatics analysis.
Taxonomic composition of samples revealed a high dominance of gamma-proteobacteria in the slaughterhouse with an average relative abundance of 75.8 ± 5.3% (Fig. 2A). Following dominant bacterial classes identified were Bacteroidia, Clostridia, Bacilli and Actinobacteria. The four generaEnhydrobacter, Moraxella, Acinetobacter and Psychrobacter, all belonging to the Moraxellaceae family, clearly dominated bacterial populations with a specific composition depending on sampling area as showed in Fig. 2B. The dehairing area was dominated by Moraxella and Acinetobacter genus. The whipping area exhibited similar bacterial composition but with high amounts of Psychrobacter for some samples. The genus Enhydrobacter clearly dominated the neck clipper area with relative abundance comprised between 52.8 and 77.7%. The carcass opener area was overall dominated by the Moraxella Genus which reached up to 91.7% of relative abundance but with one sample showing a specific composition with 6.1% of Moraxella and 27.5% of Acinetobacter. Acinetobacter and Psychrobacter dominated white offal gutter and red offal platform areas but with a higher relative abundance of Acinetobacter at red offal platform with relative abundances from 33.9 to 74.3%. In addition, analysis of bacterial population structures at the OTU (cluster) level (Fig. S1) revealed that Enhydrobacter, Moraxella or Psychrobacter populations were each dominated by only 1 or 2 recurrent OTUs for all the sampling areas. Acinetobacter genus demonstrated a higher diversity including a total of 34 different OTUs and a higher variation of OTUs proportions depending on sampling area.

Taxonomic composition of bacterial communities at the different sampling area in the slaughterhouse. For each sampling area, two bars for each of the 4 sampling dates are presented and correspond to communities before and after C&D. (A) Each bar represents relative abundances of the 5 top bacterial classes in samples. (B) Bar plots display the relative abundance of the four most abundant genera in gamma-proteobacteria. DH: dehairing, WH: whipping, NC: neck-clipper, CO: carcass opener, WOG: white offal gutter, ROP: red offal platform.
LEfSE analysis was then used to identify differentially abundant bacterial taxa between Salmonella-positive and Salmonella-negative populations (Fig. 3). Flavobacteriales, Rhodobacterales, Xanthomonadales and Aeromonadales orders were significantly more abundant in Salmonella-negative samples whereas Clostridiales and Corynebacteriales orders were more abundant in Salmonella-positive communities. Six genera including Enhydrobacter, Chryseobacterium, Aeromonas, Paracoccus, Stenotrophomonas and Soonwooa (from higher to lower LDA scores) were significantly more abundant in Salmonella-negative populations. Conversely, Acinetobacter, Hydrogenophilus, Corynebacterium, Prevotella, Flavobacterium, Lactobacillus, and Comamonas (from higher to lower LDA scores) were significantly more abundant in samples harboring Salmonella.

Bacterial taxa significantly differentiated between Salmonella-positive and Salmonella-negative populations identified by linear discriminant analysis coupled with effect size (LefSE). (A) LDA score obtained for differentially abundant taxa. Only taxa with a significant LDA threshold value of >3 are displayed. (B) Circular cladogram reporting LEfSe results presenting the identified taxa distributed according to phylogenetic characteristics.
A principal coordinate analysis (PCoA) using Unifrac and weighted Unifrac distances was performed to visualize differences in microbial population structures among samples depending on sampling areas, sampling dates or between samples before and after C&D (Fig. 4). While PERMANOVA did not show significant difference in microbial population structures among samples between sampling dates (p = 0.9905 and 0.1336 for unifrac and weighted unifrac distance respectively, Fig. 4A,B, significant effects were observed with regards to sampling area (p < 0.0001, Fig. 4C,D). Pairwise comparisons using Adonis (confirmed significant difference (p < 0.05) between all sampling areas except between WH and WOG, ROP, DH for unifrac distance and between WH andWOG, ROP, DH and CO for weighted unifrac distance, stressing the heterogeneity of WH populations depending on sampling date compared to the other sampling areas. The homogeneity of dispersion between each sampling area was checked using the betadisper function and revealed that NC area demonstrated significantly (p < 0.05) a different dispersion compared with CO and WOG for unifrac distance and only with WOG for weighted unifrac distance.

Principal coordinate analysis (PCoA) based on Unifrac and weighted Unifrac distances between samples from slaughterhouse surfaces. Samples were displayed by (A,B) sampling dates, (C,D) by sampling areas and (E,F) depending on C&D status. Adonis (PERMANOVA) statistics are also indicated. First two components explained about 41.9% and 56.4% of total variance in the dataset for Unifrac and weighted Unifrac distance matrices respectively. DH: dehairing, WH: whipping, NC: neck-clipper, CO: carcass opener, WOG: white offal gutter, ROP: red offal platform.
A significant difference was also observed between composition of bacterial populations sampled before or after C&D procedures for both unifrac (p < 0.0001 and R² = 0.23319) and weighted unifrac (p = 0.0051 and, R² = 0.0677) distances (Fig. 4). Unifrac distance PCoA (Fig. 4E) led to a better clustering of samples depending of C&D status in comparison to weighted unifrac PCoA (Fig. 4F). The comparison of α-diversity indexes (number of observed and estimated (Chao1) OTUs, Shannon and inverse Simpson indexes) showed a significant decrease for all the α-diversity index values corresponding to a decrease of richness and evenness in bacterial populations after C&D procedures (Fig. 5). To identify bacterial taxa with the greatest difference in abundance between populations before or after C&D, a LEfSE analysis using OTUs with relative abundance >1% was performed (Fig. 6). Bacterial genera more abundantly represented before (green) or after (red) C&D with LDA score and associated cladogram were displayed in Fig. 6. Numerous orders including Aeromonadales, Fusobacteriales, Clostridiales, Campylobacterales, Bacteroidales, Bacillales, Selenomonadales and Pasteurellales were more abundant in samples taken before C&D. Conversely, genera Bergeyella, Brevundimonas (and Caulobacterales order), Rothia (and Micrococcales order) and Psychrobacter were significantly more abundant (p < 0.05) in samples after C&D procedures with LDA score >3. Interestingly the significant increase of Psychrobacter relative abundance after C&D is only due to the increase in relative abundance of one dominant OTU (cluster 4 in Fig. S1).

Comparison of α-diversity indexes of microbial populations before and after C&D procedures.

Bacterial genera significantly differentiated between populations sampled before or after C&D identified by linear discriminant analysis coupled with effect size (LefSE). (A) LDA score obtained for differentially abundant genus between sample before and after C&D. Only taxa with a significant LDA threshold value of >3 are displayed. (B) Circular cladogram reporting LEfSe results presenting the identified taxa distributed according to phylogenetic characteristics.
The impact of the ethanol-based disinfection on cutting blades at NC and CO areas was also investigated using LEfSE tool. As presented on Fig. 7, Paracoccus, Moraxella and Enhydrobacter genera were statistically more abundant at these sampling areas compared to the others where no additional ethanol-based disinfection step was applied. Conversely, various genera including especially Psychrobacter and Acinetobacter for the dominant ones were less abundant at areas where additional ethanol-based disinfection was performed.

Bacterial genera significantly differentiated between bacterial populations sampled on sampling areas additionally treated with ethanol solution (NC and CO areas) identified by linear discriminant analysis coupled with effect size (LefSE). (A) LDA score obtained for differentially abundant taxa. Only genera with a significant LDA threshold value of >3 are displayed. (B) Circular cladogram reporting LEfSe results presenting the identified taxa distributed according to phylogenetic characteristics.
Discussion
Prevalence of Salmonella and associated serotypes
In the present work, Salmonella was identified at various areas in a pig slaughterhouse. Results revealed that there was no significant difference between the number of detections of Salmonella-positive samples before and after C&D procedures highlighting the ability of Salmonella to persist in the slaughterhouse environment despite C&D procedures applied. However, it should be noted that the Salmonella presence was not specifically quantified. Therefore, even if an important reduction of Salmonella levels was operated throughout the C&D procedures, it was not observable here as we investigated only the presence or absence of Salmonella after an enrichment step. Serotypes identified are common for the pig and pork sector and in agreement with previous observations34,35. We observed in particular a high prevalence of the monophasic variant of Typhimurium serotype exhibiting resistance to various antibiotics (ampicillin, tetracycline and sulfamethoxazole) which accounted for 58% of total Salmonella isolates. Such observations are consistent with its fast emergence reported since a decade and its increasing role in human infections36,37 and highlight the importance to pay specific attention to this serotype. Some PFGE-patterns were recurrently isolated at various locations in the slaughterhouse and for different dates. In one hand, our results could reflect a persistent colonization of the slaughterhouse, especially as some recurrent strains (such as B01 or B12) were also found before and after C&D for some sampling areas at a given date, underlining their ability to persist despite hygienic procedures. The potential cross-contamination of incoming pig carcasses during the slaughtering process throughout contact with Salmonella-positive surfaces in the slaughterhouse may occur at some sites and therefore represent a risk for food safety5,34,38,39. On the other hand, it could not be excluded that some strains are also highly prevalent in pigs and were thus continuously introduced in the slaughterhouse with new pig bands to be slaughtered. Strains with PFGE-patterns B04, B09 and B10 have been especially very frequently isolated in French pig slaughterhouses from meat, carcass or feces (unpublished internal data).
The fact that Salmonella was not isolated from NC area could be related to the location of this area, just after a singeing step that likely enabled the removal of Salmonella from carcasses and thus limited the contamination of the NC area. In addition, the fact that NC was also disinfected using ethanol-based solution should participate to maintain a low level of Salmonella contamination at this area. The carcass opening at the next step (CO) and the potentially associated intestinal perforation could then enable Salmonella to transfer from intestinal tract to CO surfaces, explaining why Salmonella strains were isolated at CO and then at following sampling areas. An increase of Salmonella prevalence on pig carcasses was indeed mostly observed after carcass opening and evisceration step40,41. Together, these observations again pointed out the criticality of the carcass opening step in controlling Salmonella risk at slaughterhouses as already mentioned5,42.
Microbial community diversity and link with Salmonella ecology
The sequencing and analysis of the v3-v4 region of the rDNA 16S sequences revealed that bacterial communities in the slaughterhouse were clearly dominated by the Moraxellaceae family and especially four genera: Acinetobacter, Moraxella, Psychrobacter and Enhydrobacter. From our knowledge, it is the first 16s rDNA analysis of microbial diversity from pig slaughterhouse environment, therefore there is no available data for direct comparison in the literature. Nevertheless, various published analyses of pig gut microbiome showed the dominance of Firmicutes, Bacteroidetes and Proteobacteria phyla in agreement with the data reported here43,44,45,46. Such observations are indeed coherent with the fact that bacterial populations found on the slaughterhouse surfaces are initially originating from pig carcasses and then colonize the slaughterhouse environment. In line with this observation, by evaluating the nasal microbiota composition in slaughter-aged pigs using 16s rDNA sequencing, Weese et al.47 found that Moraxella, Psychrobacter and Acinetobacter were among the most dominant genera in healthy pigs with relative abundance of 35.4, 21.1 and 4.8% respectively, in consistency with data presented here.
The site-specific composition and the stability of populations observed here over the different visits at most of the sampling areas suggest a persistent colonization of the slaughterhouse by the four dominant genera from Moraxellaceae family. These observations were supported by the analysis of composition of these genera at the OTUs level (Fig. S1) showing that only 1 or 2 persistent OTUs are found along the slaughter line and for the different sampling dates for the Psychrobacter, Enhydrobacter or Moraxella genera. Survival of bacteria in industries is mostly related to their ability to form biofilms on surfaces exhibiting high tolerance to biocidal treatments48. Therefore, it should be interesting to analyze biofilm formation and associated biocide resistance of these resident strains to better understand the mechanisms underlying their persistence in the slaughterhouse.
Analyzing the correlation between the presence of Salmonella and abundance of other bacterial taxa in surface populations, we found that some genera were significantly differentially abundant between salmonella-positive or -negative populations. Concerning dominant genera identified in the slaughterhouse, there was a clear negative correlation between Salmonella and Enhydrobacter genus. Indeed, the dominance of Enhydrobacter genus at the NC area was correlated with an absence of Salmonella for all sampling dates and interestingly Enhydrobacter genus being represented mainly by one single OTU in the slaughterhouse (Fig. S1). Therefore, it could be noteworthy to isolate this strain and test the existence of specific interactions with Salmonella using dedicated interaction assays to attribute the absence of Salmonella to a specific antagonistic interaction with the Enhydrobacter strain or only as a consequence of the singeing step applied before NC area. Reciprocally, positive correlation was found between Acinetobacter genus and Salmonella presence that may suggest positive interactions between both genera. Overall, such observations should be confirmed by repeating the analysis via a second sampling campaign, giving a view of microbial ecology in the slaughterhouse over the years.
Impact of C&D procedure on Salmonella AMR and bacterial ecology
In one hand, comparison of MIC values obtained for Salmonella strains before and after C&D procedures did not reveal significant changes in susceptibilities of isolates against the 14 antibiotics tested. These results suggested here that C&D procedures performed in the slaughterhouse did not lead to the selection of Salmonella isolates with increase resistance toward antimicrobials during the time of the study (from March to May 2017). Gantzhorn et al.49 found similar results while reporting no difference in MICs toward antibiotics nor in the number of resistances of Salmonella isolates obtained before and after C&D in six Danish pig slaughterhouses. However, by analyzing susceptibilities of Pseudomonas spp. isolated from goat and lamb slaughterhouses to biocides and antibiotics, Lavilla-Lerma et al.50 found statistical correlations between both antimicrobials revealing a co- or cross-resistance between antimicrobials especially between polyhexamethylene guanidine hydrochloride or triclosan and different antibiotics. First, the relatively short observation time of two months in the present study may not enable to observe an evolution of Salmonella antimicrobials susceptibilities. It could thus be interesting to go back in the same slaughterhouse and to sample the same areas to compare the antimicrobial resistance profiles of Salmonella strain on a longer period. Furthermore, it would be interesting to also collect data about antimicrobial resistance phenotypes of the associated dominant and persistent genera identified here in the slaughterhouse (especially Acinetobacter, Moraxella, Psychrobacter and Enhydrobacter). Indeed, these resident populations undergo repeatedly C&D procedures and are more likely to adapt in the long term. Even though they are not necessarily pathogenic strains, they can represent a potential risk throughout interactions with pathogens as they could play a role in pathogen persistence and/or constitute a potential reservoir for antimicrobial resistances for example.
On the other hand, C&D procedures applied here modified bacterial community composition and led to a loss of richness and evenness in bacterial populations as revealed by the significant decrease of all α-diversity indexes. Likewise, different studies highlighted a loss of diversity in microbial populations after exposure to various disinfectants and active substances including quaternary ammonium-based solutions51,52, chlorine53, dibromonitrilopropionamide54, and other molecules and mixed disinfectants55. Analyzing the impact of the additional ethanol-based disinfection on cutting blades, we also found that some genera as Moraxella, Enhydrobacter and Paracoccus were significantly more abundant at sampling areas treated with ethanol suggesting a potential role of ethanol disinfection step in shaping bacterial populations at NC and CO areas.
The comparison of unifrac and weighted unifrac muldimensional scaling and PERMANOVA analyses (Fig. 4E,F) showed that C&D procedures impact was overall less observable when clustering samples by integrating abundance data and not only presence/absence of OTUs. This observation illustrates that C&D rather led to an elimination of minority OTUs while it had a lower impact on population composition with regards to dominant ones. In addition, LEfSe analysis revealed that while an important number of bacterial orders indeed displayed a significant decrease of their relative abundance after C&D, few genera were favored including Bergeyella, Brevundimonas, and especially Rothia and Psychrobacter. Interestingly, Psychrobacter was also identified as one of the dominant genus in the slaughterhouse. Furthermore, one single dominant OTU (cluster 4 in Fig. S1) is responsible for the increase in relative abundance observed at all the sampling areas for Psychrobacter genus after C&D. From this observation, this strain could therefore constitute a relevant model for identifying markers of bacterial adaptation in such food environments.
Conclusion
In this contribution, original data on Salmonella prevalence and associated microbial ecology in a pig slaughterhouse were collected. The high prevalence of the monophasic variant of Typhimurium and the dominance of the Moraxellaceae family along the slaughtering chain were highlighted. Additional 16s rDNA analyses of microbial ecology should be performed over years in this slaughterhouse and also extend to a larger number of pig slaughterhouses in order to confirm such results. Complementary experiments should also be conducted to assess the existence of potential specific interactions between Salmonella and resident bacteria identified here (as inhibition with Enhydrobacter or positive interactions with Acinetobacter for instance). Interestingly, C&D procedures did not lead to the selection of Salmonella with higher resistance toward antibiotics and biocides but markedly impacted the composition of bacterial populations associated to slaughterhouse surfaces. From these observations, it seems essential to study antimicrobial resistance dynamics in industries by integrating the whole bacterial community using global approach as shotgun metagenomics. Indeed, resident flora (as Psychrobacter here) constitutes the best illustration of bacterial adaptation in a given area and could participate to the dissemination of antimicrobial resistance throughout interactions with pathogens. Overall, data collected here are important for the construction of a realistic vision of Salmonella ecology and AMR emergence in food industries with a focus on the potential role of C&D procedures.
References
EFSA & ECDC. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2016. EFSA Journal 15(12), 228 (2017).
Van Cauteren, D. et al. Estimation de la morbidité et de la mortalité liées aux infections d’origine alimentaire en France métropolitaine, 2008–2013. Bull. Epidémiol. Hebd. 1, 2–10, http://invs.santepubliquefrance.fr/beh/2018/1/2018_1_1.html (2018).
De Knegt, L. V., Pires, S. M. & Hald, T. Attributing foodborne salmonellosis in humans to animal reservoirs in the European Union using a multi-country stochastic model. Epidemiol Infect 143, 1175–1186, https://doi.org/10.1017/S0950268814001903 (2015).
Ronnqvist, M., Valttila, V., Ranta, J. & Tuominen, P. Salmonella risk to consumers via pork is related to the Salmonella prevalence in pig feed. Food Microbiol 71, 93–97, https://doi.org/10.1016/j.fm.2017.03.017 (2018).
Arguello, H., Alvarez-Ordonez, A., Carvajal, A., Rubio, P. & Prieto, M. Role of slaughtering in Salmonella spreading and control in pork production. J Food Prot 76, 899–911, https://doi.org/10.4315/0362-028X.JFP-12-404 (2013).
Møretrø, T. & Langsrud, S. Residential Bacteria on Surfaces in the Food Industry and Their Implications for Food Safety and Quality. Comprehensive Reviews in Food Science and Food Safety 16, 1022–1041, https://doi.org/10.1111/1541-4337.12283 (2017).
Giaouris, E. et al. Intra- and inter-species interactions within biofilms of important foodborne bacterial pathogens. Front Microbiol 6, 841, https://doi.org/10.3389/fmicb.2015.00841 (2015).
Habimana, O. et al. Micro ecosystems from feed industry surfaces: a survival and biofilm study of Salmonella versus host resident flora strains. BMC Vet Res 6, 48, https://doi.org/10.1186/1746-6148-6-48 (2010).
Oloso, N. et al. Antimicrobial Resistance in Food Animals and the Environment in Nigeria: A Review. International Journal of Environmental Research and Public Health 15, 1284 (2018).
Verraes, C. et al. Antimicrobial resistance in the food chain: a review. Int J Environ Res Public Health 10, 2643–2669, https://doi.org/10.3390/ijerph10072643 (2013).
Wales, A. D. & Davies, R. H. Co-Selection of Resistance to Antibiotics, Biocides and Heavy Metals, and Its Relevance to Foodborne Pathogens. Antibiotics (Basel) 4, 567–604, https://doi.org/10.3390/antibiotics4040567 (2015).
Soumet, C. et al. Reduced susceptibilities to biocides and resistance to antibiotics in food-associated bacteria following exposure to quaternary ammonium compounds. J Appl Microbiol 121, 1275–1281, https://doi.org/10.1111/jam.13247 (2016).
Webber, M. A. et al. Parallel evolutionary pathways to antibiotic resistance selected by biocide exposure. J Antimicrob Chemother 70, 2241–2248, https://doi.org/10.1093/jac/dkv109 (2015).
SCENIHR. Assessment of the Antibiotic Resistance Effects of Biocides Scientific Committee on Emerging and Newly Identified Health Risks, European commission (2009).
Soumet, C. et al. Evaluation of a multiplex PCR assay for simultaneous identification of Salmonella sp., Salmonella enteritidis and Salmonella typhimurium from environmental swabs of poultry houses. Lett Appl Microbiol 28, 113–117 (1999).
Guibourdenche, M. et al. Supplement 2003–2007 (No. 47) to the White-Kauffmann-Le Minor scheme. Res Microbiol 161, 26–29, https://doi.org/10.1016/j.resmic.2009.10.002 (2010).
Grimont P. A. D. & X., W. F. Antigenic formulae of the Salmonella serovars. Institut Pasteur & WHO Collaborating Centre for Reference and Research on Salmonella Paris, France (2007).
Ribot, E. M. et al. Standardization of pulsed-field gel electrophoresis protocols for the subtyping of Escherichia coli O157:H7, Salmonella, and Shigella for PulseNet. Foodborne Pathog Dis 3, 59–67, https://doi.org/10.1089/fpd.2006.3.59 (2006).
Hunter, S. B. et al. Establishment of a universal size standard strain for use with the PulseNet standardized pulsed-field gel electrophoresis protocols: converting the national databases to the new size standard. J Clin Microbiol 43, 1045–1050, https://doi.org/10.1128/JCM.43.3.1045-1050.2005 (2005).
Kerouanton, A. et al. Pulsed-field gel electrophoresis subtyping database for foodborne Salmonella enterica serotype discrimination. Foodborne Pathog Dis 4, 293–303, https://doi.org/10.1089/fpd.2007.0090 (2007).
Peters, T. M. et al. The Salm-gene project - a European collaboration for DNA fingerprinting for food-related salmonellosis. Euro Surveill 8, 46–50 (2003).
Barrett, T. J., Gerner-Smidt, P. & Swaminathan, B. Interpretation of pulsed-field gel electrophoresis patterns in foodborne disease investigations and surveillance. Foodborne Pathog Dis 3, 20–31, https://doi.org/10.1089/fpd.2006.3.20 (2006).
Jacouton, E., Chain, F., Sokol, H., Langella, P. & Bermudez-Humaran, L. G. Probiotic Strain Lactobacillus casei BL23 Prevents Colitis-Associated Colorectal Cancer. Front Immunol 8, 1553, https://doi.org/10.3389/fimmu.2017.01553 (2017).
Escudie, F. et al. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics 34, 1287–1294, https://doi.org/10.1093/bioinformatics/btx791 (2018).
Magoc, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963, https://doi.org/10.1093/bioinformatics/btr507 (2011).
Mahe, F., Rognes, T., Quince, C., de Vargas, C. & Dunthorn, M. Swarm: robust and fast clustering method for amplicon-based studies. PeerJ 2, e593, https://doi.org/10.7717/peerj.593 (2014).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahe, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584, https://doi.org/10.7717/peerj.2584 (2016).
Bokulich, N. A. et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10, 57–59, https://doi.org/10.1038/nmeth.2276 (2013).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73, 5261–5267, https://doi.org/10.1128/AEM.00062-07 (2007).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421, https://doi.org/10.1186/1471-2105-10-421 (2009).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–596, https://doi.org/10.1093/nar/gks1219 (2013).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217, https://doi.org/10.1371/journal.pone.0061217 (2013).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol 12, R60, https://doi.org/10.1186/gb-2011-12-6-r60 (2011).
Bonardi, S. Salmonella in the pork production chain and its impact on human health in the European Union. Epidemiol Infect 145, 1513–1526, https://doi.org/10.1017/S095026881700036X (2017).
Di Ciccio, P. et al. Microbiological contamination in Three Large-Scale Pig Slaughterhouses in Northern Italy. Ital J Food Saf 5, 6151, https://doi.org/10.4081/ijfs.2016.6151 (2016).
Hopkins, K. L. et al. Multiresistant Salmonella enterica serovar 4,[5],12:i:- in Europe: a new pandemic strain? Euro Surveill 15, 19580 (2010).
Baker, S., Thomson, N., Weill, F. X. & Holt, K. E. Genomic insights into the emergence and spread of antimicrobial-resistant bacterial pathogens. Science 360, 733–738, https://doi.org/10.1126/science.aar3777 (2018).
Hald, T., Wingstrand, A., Swanenburg, M., von Altrock, A. & Thorberg, B. M. The occurrence and epidemiology of Salmonella in European pig slaughterhouses. Epidemiol Infect 131, 1187–1203 (2003).
Piras, F., Brown, D. J., Meloni, D., Mureddu, A. & Mazzette, R. Investigation of Salmonella enterica in Sardinian slaughter pigs: prevalence, serotype and genotype characterization. Int J Food Microbiol 151, 201–209, https://doi.org/10.1016/j.ijfoodmicro.2011.08.025 (2011).
Barron, U. G. et al. Estimation of prevalence of Salmonella on pig carcasses and pork joints, using a quantitative risk assessment model aided by meta-analysis. J Food Prot 72, 274–285 (2009).
Berends, B. R., Van Knapen, F., Snijders, J. M. & Mossel, D. A. Identification and quantification of risk factors regarding Salmonella spp. on pork carcasses. Int J Food Microbiol 36, 199–206 (1997).
Letellier, A. et al. Risk factors at slaughter associated with presence of Salmonella on hog carcasses in Canada. J Food Prot 72, 2326–2331 (2009).
Kim, H. B. & Isaacson, R. E. The pig gut microbial diversity: Understanding the pig gut microbial ecology through the next generation high throughput sequencing. Veterinary Microbiology 177, 242–251, https://doi.org/10.1016/j.vetmic.2015.03.014 (2015).
Quan, J. et al. A global comparison of the microbiome compositions of three gut locations in commercial pigs with extreme feed conversion ratios. Scientific Reports 8, 4536, https://doi.org/10.1038/s41598-018-22692-0 (2018).
Holman, D. B., Brunelle, B. W., Trachsel, J. & Allen, H. K. Meta-analysis To Define a Core Microbiota in the Swine Gut. mSystems 2, https://doi.org/10.1128/mSystems.00004-17 (2017).
Crespo-Piazuelo, D. et al. Characterization of bacterial microbiota compositions along the intestinal tract in pigs and their interactions and functions. Sci Rep 8, 12727, https://doi.org/10.1038/s41598-018-30932-6 (2018).
Weese, J. S., Slifierz, M., Jalali, M. & Friendship, R. Evaluation of the nasal microbiota in slaughter-age pigs and the impact on nasal methicillin-resistant Staphylococcus aureus (MRSA) carriage. BMC Vet Res 10, 69, https://doi.org/10.1186/1746-6148-10-69 (2014).
Bridier, A. et al. Biofilm-associated persistence of food-borne pathogens. Food Microbiol 45, 167–178, https://doi.org/10.1016/j.fm.2014.04.015 (2015).
Gantzhorn, M. R., Pedersen, K., Olsen, J. E. & Thomsen, L. E. Biocide and antibiotic susceptibility of Salmonella isolates obtained before and after cleaning at six Danish pig slaughterhouses. Int J Food Microbiol 181, 53–59, https://doi.org/10.1016/j.ijfoodmicro.2014.04.021 (2014).
Lavilla Lerma, L., Benomar, N., Casado Munoz Mdel, C., Galvez, A. & Abriouel, H. Correlation between antibiotic and biocide resistance in mesophilic and psychrotrophic Pseudomonas spp. isolated from slaughterhouse surfaces throughout meat chain production. Food Microbiol 51, 33–44, https://doi.org/10.1016/j.fm.2015.04.010 (2015).
Costa, D. et al. Occurrence and diversity of both bacterial and fungal communities in dental unit waterlines subjected to disinfectants. Pathog Dis 74, https://doi.org/10.1093/femspd/ftw094 (2016).
Forbes, S. et al. Formulation of Biocides Increases Antimicrobial Potency and Mitigates the Enrichment of Nonsusceptible Bacteria in Multispecies Biofilms. Appl Environ Microbiol 83, https://doi.org/10.1128/AEM.03054-16 (2017).
Bertelli, C. et al. Reduced Chlorine in Drinking Water Distribution Systems Impacts Bacterial Biodiversity in Biofilms. Frontiers in Microbiology 9, https://doi.org/10.3389/fmicb.2018.02520 (2018).
Reynolds-Clausen, K., Surridge-Talbot, K., Botes, M. & Eugene Cloete, T. Bacterial species diversity as an indicator of dibromonitrilopropionamide (DBNPA) biocide efficacy. Water Science and Technology 78, 320–328, https://doi.org/10.2166/wst.2018.289 (2018).
Jiang, L. et al. Effect of Different Disinfectants on Bacterial Aerosol Diversity in Poultry Houses. Front Microbiol 9, 2113, https://doi.org/10.3389/fmicb.2018.02113 (2018).
Acknowledgements
This work was supported in the frame of the EcoAntibio plan funded by the French ministry for food and agriculture. We thank GeT-PlaGe Genotoul platform for performing the 16S MiSeq sequencing.
Author information
Authors and Affiliations
Contributions
A.B., A.L.R., C.F. and C.S. collected samples. A.B., A.L.R., C.F. and C.S. conceived and designed the experiments. A.B., P.L.G., M.H.M. and C.P. performed the experiments. A.B., A.L.R., C.F. and C.S. analyzed the data. A.B. prepared the manuscript. A.B., A.L.R., C.F. and C.S. reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bridier, A., Le Grandois, P., Moreau, MH. et al. Impact of cleaning and disinfection procedures on microbial ecology and Salmonella antimicrobial resistance in a pig slaughterhouse. Sci Rep 9, 12947 (2019). https://doi.org/10.1038/s41598-019-49464-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49464-8
This article is cited by
-
Occurrence, genetic diversity and resistance profiles of Salmonella enterica from Brazilian sausages collected at production facilities
Journal of Food Science and Technology (2024)
-
Comparative study on inhibitory effects of ferulic acid and p-coumaric acid on Salmonella Enteritidis biofilm formation
World Journal of Microbiology and Biotechnology (2022)
-
The sources and transmission routes of microbial populations throughout a meat processing facility
npj Biofilms and Microbiomes (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
