
Abstract
Nonnative, invasive feral pigs (Sus scrofa) modify habitats by disturbing soils and vegetation, which can alter biogeochemical processes. Soil microbial communities drive nutrient cycling and therefore also play important roles in shaping ecosystem structure and function, but the responses of soil microbes to nonnative ungulate removal remains poorly studied. We examined changes in the soil bacterial community over a ~25 year chronosequence of feral pig removal in tropical montane wet forests on the Island of Hawai’i. We extracted bacterial eDNA from soil samples collected inside and outside of ungulate exclosures along this chronosequence and sequenced the eDNA using the Illumina platform. We found that ungulate removal increased diversity of soil bacteria, with diversity scores positively correlated with time since removal. While functional and phylogenetic diversity were not significantly different between pig present and pig removed soils, soil bulk density, which decreases following the removal of feral pigs, was a useful predictor of dissimilarity among sites and correlated to changes in functional diversity. Additionally, increases in soil porosity, potassium, and calcium were correlated to increases in functional diversity. Finally, sites with greater mean annual temperatures were shown to have higher scores of both functional and phylogenetic diversity. As such, we conclude that feral pigs influence overall bacterial community diversity directly while influencing functional diversity indirectly through alterations to soil structure and nutrients. Comparatively, phylogenetic differences between communities are better explained by mean annual temperature as a climatic predictor of community dissimilarity.
Similar content being viewed by others

Introduction
Soil microbes play critical roles in water retention and purification, nutrient cycling, primary production, soil formation and processing, and carbon sequestration, with individual taxa capable of contributing either highly specialized or generalist roles1. As scientists have begun to recognize the important roles microbes play in these processes, studies examining the soil microbiome have become increasingly common. As a result, numerous factors including pH, soil water content2,3, cation exchange capacity3, and soil organic matter content4 have been shown to influence the composition, structure, and function of soil microbial communities.
Given that many soil microbes are highly sensitive to these localized environmental factors, the soil microbiome has been associated with both above- and belowground plant and animal communities1,2,5–7. For example, microbial biomass and respiration have been shown to increase with increases in plant species richness8, and the microbiome in another study was more diverse under native tree canopies than under canopies of non-native plants9. Further, the global beta-diversity of grassland plants has been strongly correlated to the beta-diversity of soil microbial communities10. The removal of ungulate grazing was also shown to decrease biodiversity in grasslands11,12 and reduce microbial activity in a forest ecosystem6.
In consideration of the potential associations between ungulate and microbial communities, the effects of specific ungulates, such as invasive feral pigs (Sus scrofa), on aboveground environments are often readily visible and can translate into indirect effects on the belowground community of microorganisms13. Prior research has shown that feral pigs can influence nearly all aspects of their habitat14,15,16. Resultant changes include decreased diversity and abundance of plants17,18, fungi19, and wildlife20,21 as well as being associated with the increased presence of specific bacteria in watersheds22,23,24. Comparatively, the effects of feral pigs on soils are typically highly variable, due to the narrow, localized effects of their trampling, wallowing, and rooting behaviors. At the level of individual study plots, examinations of feral pig influences on soils have shown mixed results. For example, in Tennessee’s deciduous forests, feral pig activity reduced soil bulk density (SBD)25, while SBD increased with feral pig activity in both Spanish alpine habitats26 and Hawaiian tropical montane wet forests27. Physical alteration of soil organic matter via foraging was also documented in some28,29,30, but not all ecosystems where it has been examined31. Moreover, feral pigs also influence soil nutrient cycling and availability, with several studies documenting increased soil nitrogen (N) availability in the presence of feral pigs26,29,30.
Feral pigs have also been associated with direct effects on soil microbial communities. Specifically, feral pigs have been shown to affect the soil microbial community through defecation6 and soil turbation32 resulting in alterations specific to the composition of soil microbial communities33. For example, feral pigs have been associated with an increased presence of fecal coliform in watersheds22 and have also been shown to increase the presence of specific bacteria, such as enterococci and leptospira, in soil runoff23,24,34. Feral pig activity has been directly associated with increased soil microbial activity attributed to potential increases of soil N availability associated with feces and urine6,35 and indirectly via increased availability of soil organic matter36. Further, the presence of feral pigs has been associated with faster microbial community recovery following rapid unexpected changes in the soil environment, such as the application of fumigants37.
In Hawai’i, and throughout much the Pacific Island region, feral pigs serve an important role culturally, but have been associated with many negative alterations to native ecosystems. As a result, land managers primarily utilize exclosure fencing coupled with removal as the primary non-lethal management strategy for feral pigs in Hawai’i16. Prior research examining the outcomes of this management strategy have indicated that the removal of feral pigs from Hawaiian tropical montane wet forests increases understory plant species richness and density, increases the cycling and availability of soil N, and decreases SBD and soil aggregation17,18,27. In Hawai’i, examinations of microbial changes following feral pig removal have been limited to short-term (<2 years) examinations of individual taxa and did not analyze changes to the entire community22,23,24. Additionally, long-term studies of ungulate removal occurring globally have been largely limited to grassland systems11,12. As such, our goal was to examine the effects of feral pig removal on soil microbial communities across a 25-year chronosequence of removal in a forest ecosystem in Hawai’i. We asked the following questions: (1) How does the removal of feral pigs affect community and functional diversity of soil bacteria?; (2) Do these effects change with time since pig removal?; and (3) How do differences in environmental variables (e.g. climatic, floral, and soil variation; Table 1) affect the community and functional diversity of soil bacteria?
Given the potential for direct effects of feral pigs on the bacterial community, we hypothesized that feral pig removal would result in a less diverse soil bacterial community, likely due to the loss of nutrient inputs associated with feral pigs including urine and feces deposition, mirroring prior long-term removal work completed in grassland ecosystems11,12. We therefore additionally hypothesized that functional and phylogenetic diversity would decrease following feral pig removal due to the loss of groups associated with these nutrient pulses. We further hypothesized that bacterial diversity would not change over time following the extended removal of feral pigs due to the ability of soil bacterial communities to rapidly adapt to environmental changes17,38,39, which we surmised would outweigh characteristics of the chronosequence shown to change more slowly by prior work18,27. In regards to the influence of environmental factors (Table 1), we hypothesized that: (A) these variables would correlate to differences in the bacterial community, but would be less predictive of community dissimilarities than would the presence or removal of feral pigs; and (B) variables indirectly associated with feral pig removal (e.g. SBD and understory plant composition) would be more influential on changes to the bacterial community than environmental variables not associated with feral pigs (e.g. mean annual temperature (MAT) and elevation). These two hypotheses are driven by the understanding that feral pigs are highly influential on community composition in this system16,18.
Results
Across all samples, eDNA extraction and Illumina MiSeq sequencing resulted in 53,140 to 81,823 raw reads per sample with qubit quantification results ranging from 42.0–85.6 ng/uL. Subsequent quality control and delineation of operational taxonomic units (OTUs) with QIIME 2 resulted in 8,503 total OTUs being included in our analyses (Appendix 1). PICRUSt analysis further organized these OTUs into 328 functional groups (Appendix 2).
Our initial assessment of the effects of environmental variables on bacterial community dissimilarity indicated that mean annual temperature (MAT), elevation, SBD, soil porosity, soil potassium (K), soil calcium (Ca), and the sum of stem density of large trees, small trees and shrubs, and tree ferns were all potential predictors of variation among bacterial communities at each of the nine sites. Following this assessment, comparing metrics of biodiversity (Shannon biodiversity index, rarified richness, Faith’s phylogenetic distance, functional Shannon biodiversity index, and functional rarefied richness) with the study site variables deemed as useful predictors led to three primary results. First, only the removal of feral pigs was associated with changes to overall bacterial community biodiversity. Specifically, the removal of feral pigs resulted in higher Shannon diversity scores (n = 9, Σ2 = 4.86, p = 0.03; Fig. 1), which was mirrored by results showing an increase in Shannon diversity scores over time following removal (n = 6, F = 14.81, p = 0.02, r2 = 0.73; Fig. 2). However, the removal of feral pigs did not directly result in any changes to either the functional (n = 9, Σ2 = 2.16, p = 0.14) or phylogenetic composition of bacterial communities (n = 9, Σ2 = 0.54, p = 0.46).

Shannon diversity in comparison to the removal/presence of feral pigs. Sites were categorized into those with feral pigs removed (in blue) and sites with feral pigs present (in red). The mean Shannon diversity score for these sites indicates that soil bacterial communities are more diverse following the removal of feral pigs (n = 9, Σ2 = 4.86, p = 0.03).

Changes in Shannon diversity score following time since feral pig removal. Following the removal of feral pigs, we see a positive linear association in the Shannon diversity of soil bacterial communities (n = 6, F = 14.81, p = 0.02, r2 = 0.73). Along the left side of the figure at time = 0, the red circle represents the mean of the four sites with feral pigs still present all of which do not have removal. The remaining five points symbolized with blue boxes represent each of the five plots with feral pigs removed. Error bars represent standard deviation.
Instead, our second primary result indicated that environmental characteristics were the principle variables explaining variation in functional community structure across our study sites. Individually, Shannon diversity scores of functional groups were specifically associated with SBD (n = 9, F = 18.37, p = 0.004, r2 = 0.68), soil porosity (n = 9, F = 17.55, p = 0.004, r2 = 0.67), soil K (n = 9, F = 13.80, p = 0.008, r2 = 0.62), and soil Ca (n = 9, F = 6.13, p = 0.04, r2 = 0.39). Comparatively, the functional richness of the bacterial community at each site was associated with MAT (n = 9, F = 32.82, p < 0.001, r2 = 0.80), elevation (n = 9, F = 37.05, p < 0.001, r2 = 0.82), SBD (n = 9, F = 8.70, p = 0.02, r2 = 0.49), soil porosity (n = 9, F = 8.58, p = 0.02, r2 = 0.49), soil K (n = 9, F = 37.37, p < 0.001, r2 = 0.82), and soil Ca (n = 9, F = 29.33, p < 0.001, r2 = 0.78).
Our third primary result indicated that only two environmental variables were associated with changes in the phylogenetic diversity of the bacterial communities at each site: MAT (n = 9, F = 10.00, p = 0.02, r2 = 0.53; Fig. 3) and elevation (n = 9, F = 8.10, p = 0.02, r2 = 0.47). In these cases, small changes in MAT (~1.5 °C) were associated with increased Faith’s phylogenetic diversity scores while increases in elevation (~200 m) were associated with decreased diversity scores. Importantly, MAT and elevation are linearly and inversely correlated in this system. The remaining environmental variables tested (including geographic distance between sites using principle coordinates of neighbor matrices (PCNM), size of removal units, mean annual precipitation (MAP), ground covered by litter, ground covered by bryophytes, count of terrestrial seedlings, water-filled pore space, soil magnesium (Mg), and the sum of basal area of large trees, small trees and shrubs, and tree ferns) were not significantly associated with any biodiversity metrics.

Correlating mean annual temperature (MAT) to Faith’s phylogenetic diversity. Faith’s phylogenetic diversity of the bacterial communities (n = 9, F = 10.00, p = 0.02, r2 = 0.53) was positively and linearly associated with MAT. Red circles represent sites with pigs present and blue boxes are sites where pigs have been removed. Error bars signify standard deviation.
The results of our non-metric multidimensional scaling (NMDS) analysis indicated that the presence or removal of feral pigs (p = 0.01, r2 = 0.99; Fig. 4A), MAT (p = 0.003, r2 = 0.93; Fig. 4B), and SBD (p = 0.01, r2 = 0.79; Fig. 4C) were the only significant predictors of bacterial community dissimilarity between sites in the simplified model. None of the other variables associated with changes in biodiversity metrics (time since feral pig removal, elevation, soil porosity, soil K, soil Ca, and the sum of stem density of large trees, small trees and shrubs, and tree ferns) were significant predictors of bacterial community dissimilarity.

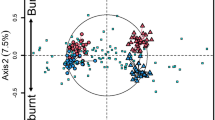
(A–C) Explaining bacterial community dissimilarity. Dissimilarities among bacterial communities were scored along two axes established by differences in the frequencies of individual operational taxonomic units (OTUs) at each site and placed into a dissimilarity index, displayed here. Each of the two non-metric multidimensional scaling (NMDS) axes describes dissimilarity between communities along an arbitrary scale; these two scales are the x and y axes of this figure. Within the figure, the circles represent the bacterial community at the individual sites in our study; blue circles have feral pigs present and red circles represent sites with feral pigs removed. The location of sites in the scaling space is the same across all three figures. In Fig. 4A, the purple sub-axis labeled “Pigs” represents the presence/absence of feral pigs at each site, and the horizontal lines within the figure show the scale of this axis where 1 represents sites with pigs present and 0 represents sites with pigs absent. This presence/absence is a good predictor of the dissimilarity among the bacterial communities (p = 0.004, r2 = 0.99). In Fig. 4B, the purple sub-axis labeled “MAT” represents mean annual temperature (MAT) at each site. The vertical lines within the figure scale the MAT from 14.5–15.9 °C. MAT was a significant predictor of dissimilarity among the bacterial communities (p = 0.003 r2 = 0.93). In Fig. 4C, the purple sub-axis labeled “SBD” represents soil bulk density (SBD) at each site. The vertical lines within the figure scale the SBD from 0.19–0.53 g/cm3. SBD was a significant predictor of dissimilarity among the bacterial communities (p = 0.01, r2 = 0.79).
Discussion
We originally hypothesized that the diversity of soil bacterial communities would decrease following the removal of feral pigs and would not vary over time following the removal of feral pigs, with functional and phylogenetic diversity following a similar pattern. Additionally, we hypothesized that the removal of feral pigs as a direct effect would better predict dissimilarities among soil bacterial communities than the indirect and unrelated effects of individual environmental variables. Our results, however, did not support these hypotheses. Instead, the diversity of soil bacterial communities was significantly higher in sites where feral pigs had been removed compared to unfenced sites, and diversity increased positively and linearly with time since the removal of feral pigs. Further, neither functional nor phylogenetic metrics of diversity showed overall increases associated with time since removal. Rather, functional diversity and phylogenetic diversity remained relatively constant in response to feral pig removal, as well as over time following removal. Finally, both MAT and SBD were also identified as useful predictors of bacterial community dissimilarity in addition to the removal of feral pigs. Changes in SBD have been previously associated with the removal of feral pigs in our study system27 and can be viewed as an indirect effect of feral pig removal, and differences associated with MAT are unrelated to effects by feral pigs.
In consideration of the direct effects of feral pigs on the soil bacterial community, prior research has indicated that feral pig activity (including rooting, trampling, etc.) leads to positive increases in abundance and activity of soil microbes6,35,36. Further, the long-term removal of ungulates from grassland systems resulted in decreased soil bacterial diversity11,12 leading us to hypothesize that soil bacterial diversity would decrease following the removal of feral pigs. However, the results of our study directly conflict with these prior results despite similar trends of above- and belowground recovery observed in our study system and the system examined by Wang, et al.12. Specifically, in both systems, plant cover, plant diversity, and soil nutrient levels all increased following the exclusion of ungulates. A likely explanation for this dichotomy is the stark contrast between the two study systems being a grassland and a tropical wet forest. However, this does not mechanistically explain the differences we observed, and our study is limited in its ability to discern these differences.
While we are unable to explain a causative agent for the difference observed between our study and Wang, et al.12, we can speak to the some of the indirect effects of ungulate removal on the bacterial community in our own system. In our study, we identified SBD as a useful predictor of dissimilarity among soil bacterial communities, with lower values of SBD correlating to increased bacterial diversity. Additionally, increased levels of soil porosity, K, and Ca were all also associated with increased levels of functional diversity. In order to understand these effects as indirectly related to feral pigs, it is important to note that prior research in our study system has indicated that feral pig removal resulted in a significant decrease in SBD as well as significant increases in soil porosity, K, and Ca27. As such, changes to the soil bacterial community associated with these variables can largely be attributed to indirect effects of feral pig removal. These results are supported by prior work indicating that soil characteristics can explain changes to the soil microbial community2,3,4, but in our study, changes associated with soil characteristics can be indirectly attributed to feral pigs.
Our results also indicate that changes in MAT and elevation both correlate to changes in the functional and phylogenetic diversity of soil bacterial communities. Naturally, neither of these variables are affected by feral pigs, and they are inversely and linearly correlated to one another. In our analysis of these variables, MAT was a good predictor of dissimilarity among bacterial communities. This result was exemplified by the increased functional and phylogenetic diversity at warmer sites, despite small changes in MAT (~1.5 °C), thereby suggesting that, while feral pig removal does result in changes to the soil bacterial diversity, overall community structure is more sensitive to fine-scale differences in environmental variables (i.e., MAT). This result aligns with previous work showing that soil microbial communities are highly sensitive to environmental factors at small scales3,9. Specifically, studies conducted worldwide have indicated that temperature plays a critical role in the diversity and composition of soil microbial communities40,41 and has been supported by recent work suggesting microbes follow Humboldt’s patterns of tropical plant species richness, decreasing in correlation to decreased temperature and increased elevation across a much larger gradient than used in our study42. However, it is important to note that our results contradict those previously reported by another field study conducted in Hawai’i suggesting that soil microbial communities are only minimally affected by changes in MAT2. This contradiction may indicate that some other variable, such as soil nitrogen, may be influenced by MAT43 in our system resulting in changes to the soil bacterial community across a small gradient that were not captured by our study.
The results of this study provide important information for understanding changes over time in soil bacterial communities following the removal of nonnative ungulates, an increasingly common management approach globally. In this study, we observed a linear increase in soil bacterial diversity over time following feral pig removal that did not directly correlate to any increases in functional or phylogenetic diversity. This result indicates increases in functional overlap over time, which for some communities can increase ecosystem stability43. In our study area, the soil attributes we observed influencing these changes in functional diversity (including SBD, soil porosity, soil K, and soil Ca) were previously identified as changing following the removal of feral pigs27. As such, we can attribute the increase in functional overlap indirectly to the removal of feral pigs. Comparatively, phylogenetic differences between bacterial communities were only associated with variables that are not influenced by feral pigs (i.e. MAT and elevation) leading us to conclude that the phylogenetic structure of bacterial communities at our study site is not influenced, directly nor indirectly, by feral pigs. In the future, it is likely that a more in-depth functional metagenomic analysis could improve our understanding of this system.
Methods
Study site
This study utilized a chronosequence of feral pig removals on the Island of Hawai’i initially characterized by Cole and Litton18. The experimental design consists of five pairs of sites arrayed across feral pig removal units located on the eastern side of Mauna Loa Volcano in the Ola’a Unit of Hawai’i Volcanoes National Park and the adjacent Pu’u Maka’ala Natural Area Reserve. These pairs include four sites with feral pigs present and five sites with feral pigs removed (with one pig present site serving as the pair for two pig removal sites) at intervals ranging from ~13 to ~25 years prior to our study18. One limitation of this study is the lack of spatial replicates, and our results should therefore be interpreted with appropriate caution. However, this system is the best available for analyzing differences in wet forest ecosystems because there are no longer any naturally occurring pig-free areas within these habitats in Hawai’i, and the cost of constructing these units is high due to their remote location and rugged terrain. Combined, these factors inherently limit the study of these processes in regards to feral pigs. Further, these removal units range in size from 117–1,024 ha providing enough geographic space for spatial replicates, but doing so would result in the pseudoreplication of data.
All study sites occur on 2,000–10,000 year-old tephra-derived andisols that are characterized by deep, moderately well-drained soils from basic volcanic ash deposited over basic lava. These soils stem from two closely related soil series: Puaulu (medial over ashy, aniso, ferrihydritic over amorphic, isothermic Aquic Hapludands) and Eheuiki (medial, ferrihydritic, isothermic Typic Hydrudands) with slopes of 2–5%18,27,44. Sites range in elevation from 1,140–1,370 m above sea level, with MAT inversely correlating to elevation and covering a range of 14.4–15.9 °C. MAP is between 2,910–3,985 mm at the sites with no distinct seasonality45. Vegetation is characterized by large areas of canopy-intact native tropical montane wet forests classified as Metrosideros polymorpha/Cibotium spp.46. Feral pig densities have been estimated at 0.6–16.3 feral pigs/km2 at these sites47, and excluding recreational hunting, forests surrounding the exclosures have never been managed for feral pigs18,27. Significant prior work has been conducted in these sites and the information from these studies, which we have utilized for the sake of our own analyses, is summarized in Table 1; however, this is not an exhaustive list of all variables that could possibly be studied within this ecosystem.
Soil collection
During May of 2017, three soil samples were collected from unique plots within each of the nine study sites described above for a total of 27 samples. In order to control for canopy cover and associated aboveground characteristics in the local environment, the center of each plot was placed within 1 m of a mature tree fern (Cibotium spp.). Each sample was collected by gathering eight subsamples from 1 and 3 m intervals in the four cardinal directions from each plot’s center. Leaf litter was cleared to expose Oa- and A-horizon soils, and a clean spatula, latex gloves, and Falcon tube were used to collect each soil subsample (<12 mL in volume) from the top 1–2 cm of the soil from each of the eight subsample points. Once the subsamples were collected and sealed into a single Falcon tube representing the entire sample, the sample was shaken by hand to homogenize the soil and placed in a cooler for ~3–7 hours until it could be transferred to a −20.0 °C freezer for longer-term storage until analysis.
eDNA sequencing and analysis
To assess the soil bacterial community in each subplot, eDNA was extracted from the soil samples using MP Biomedicals’ FastDNA SPIN Kit for Soil (MP Biomedicals, Santa Anna, California, USA). Manufacturer provided protocols were followed, including extended centrifugation at 14,000 rpm to remove excessive debris and an extra 5 minutes of incubation at 55 °C to increase eDNA yields. The standard Illumina 16S Metagenomic Sequencing Library Preparation workflow was used to prepare samples for sequencing the 464 bp variable V3 and V4 regions of the 16S rRNA gene48 using the primer pairs forward S-D-Bact-0341-b-S-17 (5′-CCTACGGGNGGCWGCAG-3′) and reverse S-D-Bact-0785-a-A-21 (5′-GACTACHVGGGTATCTAATCC-3′)49. These primers were concatenated to standard Illumina adapters, annealed to eDNA sample extracts at 95 °C for 3 minutes, and amplified using KAPA HiFi HotStart ReadyMix for 25 cycles at 95 °C for 30 seconds, 55 °C for 30 seconds, and 72 °C for 30 seconds and finally held at 72 °C for 5 minutes48. Amplified products were purified twice using AMPure XP magnetic beads (Beckman Coulter, Brea, California, USA) and quantified using Biotium’s AccuBlue High Sensitivity dsDNA Solution Kit on a Qubit 3.0 fluorometer per manufacturer guidelines (Biotium, Fremont, California, USA). Libraries were combined at equimolar concentration and sequenced at the University of Hawai’i at Mānoa Advanced Studies in Genomics, Proteomics, and Bioinformatics (ASGPB) genomics core facility on a 300 bp paired-end Illumina MiSeq platform. All samples were multiplexed using the Illumina NextEra XT index kit. Negative controls did not result in any sequences after processing and were thereafter removed from our analyses. Raw sequence reads were deposited in MG-RAST (Project # 87547).
Molecular sequences were processed using the QIIME 2 bioinformatics platform version 2018.4 within a Virtual Box Core50. Initial sequences were demultiplexed and truncated at the 10th bp from the left for both the forward and reverse reads. From the right, the sequences were truncated from the 290th bp on the forward read and from the 250th bp on the reverse read. Divisive Amplicon Denoising Algorithm (DADA2) was then used to merge paired reads, filter by sequence quality, denoise, and create a sample × OTU table, and remove chimeras51. OTUs were then taxonomically assigned using the Naïve Bayes Classifier trained on the Greengenes 13_8 99% OTU database. OTUs occurring <10 times across all 27 samples were removed. The final OTU table was rarefied to 8,503 sequences and used for all subsequent analyses (Appendix 1). Secondarily, OTUs were assigned to functional groups using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) bioinformatics software51. This step was completed with an open reference search within the QIIME 2 platform of the Greengenes 13_5 database. The resultant file listed the predicted functional groups based-off specific 16S rRNA OTUs52 (Appendix 2). These functional groups are established by placing each OTU into a functional category determined by their phylogeny and related information in the Greengenes database. QIIME 2 was then used to obtain biodiversity scores: Shannon biodiversity index, rarified richness, and Faith’s phylogenetic distance as well as Shannon biodiversity index and rarefied richness describing predicted functional diversity.
Statistical analyses
Data from each of the 27 total samples collected was averaged across the three samples from each site resulting in statistical analyses of the nine sites existing in the chronosequence (n = 9). Statistical analyses were then conducted using R-Studio Version 3.3.453. Biodiversity scores obtained in QIIME 2 were compared against available site characteristics obtained from Cole and Litton18, Long, et al.27, and Giambelluca et al.45 summarized in Table 1. Due to the non-parametric nature of the data, Kruskal-Wallis rank sum tests were used to determine if any differences in biodiversity were associated with categorical site characteristics including presence/absence of feral pigs (Fig. 1) and soil series. Comparatively, continuous site characteristics (time since removal, MAP, MAT, elevation, etc.) were compared in relation to biodiversity metrics using linear regression (Figs. 2 and 3).
In addition to comparisons of biodiversity metrics, NMDS vector fitting was used to fit site characteristics and comparisons of Euclidean distance was completed using principle coordinates of neighbor matrices (PCNM). These analyses were conducted using the <vegan> package available within R54. For each subset, basic ordination of community structure was established at each site using linear directional gradients <envfit>. These vectors were then plotted with surface fitting for improved clarity of nonlinear vector relationships <ordisurf>. Finally, constrained correspondence analysis (CCA) was used to determine site characteristics significantly affecting community composition dissimilarities to 1,000 iterations. Permutation tests were then conducted to select the best fitting model (Fig. 4A–C).
Data Availability
Raw data is identified in the manuscript as Appendices 1 & 2 and have been submitted with the manuscript. Additionally, raw genetic sequences are publicly availableas MG-Rast Project #87547.
References
Lavelle, P. et al. Soil invertebrates and ecosystem services. Eur. J. Soil Biol. 42, S3–S15 (2006).
Selmants, P. C., Adair, K. L., Litton, C. M., Giardina, C. P. & Schwartz, E. Increases in mean annual temperature do not alter soil bacterial community structure in tropical montane wet forests. Ecosphere 7, e01296, https://doi.org/10.1002/ecs2.1296 (2016).
Docherty, K. M. et al. Key edaphic properties largely explain temporal and geographic variation in soil microbial communities across four biomes. PLoS One 10, e0135352, https://doi.org/10.1371/journal.pone.0135352 (2015).
King, G. M. Contributions of atmospheric CO and hydrogen uptake to microbial dynamics on recent Hawaiian volcanic deposits. Appl. Environ. Microbiol. 69, 4067–4075 (2003).
Kao-Kniffin, J. & Balser, T. C. Soil fertility and the impact of exotic invasion on microbial communities in hawaiian forests. Microb. Ecol. 56, 55–63 (2008).
Mohr, D., Cohnstaedt, L. W. & Topp, W. Wild boar and red deer affect soil nutrients and soil biota in steep oak stands of the Eifel. Soil Biol. Biochem. 37, 693–700 (2005).
Duhamel, M. et al. Plant selection initiates alternative successional trajectories in the soil microbial communitiy after disturbance. Ecological Monographs 89, e01367 (2019).
Zak, D. R., Holmes, W. E., White, D. C., Peacock, A. D. & Tilman, D. Plant diversity, soil microbial communities, and ecosystem function: are there any links? Ecology 84, 2042–2050 (2003).
Lynch, H. B., Epps, K. Y., Fukami, T. & Vitousek, P. M. Introduced canopy tree species effect on the soil microbial community in a montane tropical forest. Pac. Sci. 66, 141–150 (2012).
Prober, S. M. et al. Plant diversity predicts beta but not alpha diversity of soil microbes across grasslands worldwide. Ecol. Lett. 18, 85–95 (2015).
Cheng, J., Jing, G., Wei, L. & Jing, Z. Long-term grazing exclusion effects on vegetation characteristics, soil properties and bacterial communities in the semi-arid grasslands of China. Ecol. Eng. 97, 170–178 (2016).
Wang, Z. et al. Impact of long-term grazing exclusion on soil microbial community composition and nutrient availability. Biol. Fert. Soils 55, 121–134 (2019).
Wardle, D. A. et al. Ecological linkages between aboveground and belowground biota. Science 304, 1629–1633 (2004).
Campbell, T. A. & Long, D. B. Feral swine damage and damage management in forested ecosystems. Forest Ecol. Manag. 257, 2319–2326 (2009).
Barrios-Garcia, M. & Ballari, S. Impact of wild boar (Sus scrofa) in its introduced and native range: a review. Biol. Invasions 14, 2283–2300 (2012).
Wehr, N. H., Hess, S. C. & Litton, C. M. Biology and impacts of Pacific Islands invasive species. 14. Sus scrofa, the feral pig (Artiodactyla: Suidae). Pac. Sci. 72, 177–198 (2018).
Cole, R. J., Litton, C. M., Koontz, M. J. & Loh, R. K. Vegetation recovery 16 years after feral pig removal from a wet Hawaiian forest. Biotropica 44, 463–471, https://doi.org/10.1111/j.1744-7429.2011.00841.x (2012).
Cole, R. J. & Litton, C. M. Vegetation response to removal of non-native feral pigs from Hawaiian tropical montane wet forest. Biol. Invasions 16, 125–140 (2014).
Soteras, F. et al. Mycophagy by invasive wild boar (Sus scrofa) facilitates dispersal of native and introduced mycorrhizal fungi in Patagonia, Argentina. Fungal Ecol. 26, 51–58 (2017).
Atkinson, C. T. & LaPointe, D. A. Introduced avian diseases, climate change, and the future of Hawaiian honeycreepers. J. Avian Med. Surg. 23, 53–63 (2009).
Galetti, M. et al. Diet overlap and foraging activity between feral pigs and native peccaries in the pantanal. PLoS One 10, e0141459, https://doi.org/10.1371/journal.pone.0141459 (2015).
Strauch, A. M., Bruland, G. L., MacKenzie, R. A. & Giardina, C. P. Soil and hydrological responses to wild pig (Sus scofa) exclusion from native and strawberry guava (Psidium cattleianum)-invaded tropical montane wet forests. Geoderma 279, 53–60 (2016).
Dunkell, D. O., Bruland, G. L., Evensen, C. I. & Walker, M. J. Effects of feral pig (Sus scrofa) exclusion on Enterococci in runoff from the forested headwaters of a Hawaiian watershed. Water Air Soil Poll. 221, 313–326 (2011).
Bovino-Agostini, V., Bruland, G. L., Litton, C. M. & Evensen, C. I. Spatial and temporal dynamics and effects of feral pigs (Sus scrofa) on Enterococci in soil and runoff of a forested watershed in Hawaii. Thesis at, https://scholarspace.manoa.hawaii.edu/handle/10125/101404 (2012).
Singer, F. J., Swank, W. T. & Clebsch, E. E. C. Effects of wild pig rooting in a deciduous forest. J. Wildlife Manage. 48, 464–473 (1984).
Bueno, C. G., Azorín, J., Gómez-García, D., Alados, C. L. & Badía, D. Occurrence and intensity of wild boar disturbances, effects on the physical and chemical soil properties of alpine grasslands. Plant Soil 373, 243–256 (2013).
Long, M. S. et al. Impact of nonnative feral pig removal on soil structure and nutrient availability in Hawaiian tropical montane wet forests. Biol. Invasions 19, 749–763 (2017).
Hobbs, N. T. Modification of ecosystems by ungulates. J. Wildlife Manage. 60, 695–713 (1996).
Siemann, E., Carrillo, J. A., Gabler, C. A., Zipp, R. & Rogers, W. E. Experimental test of the impacts of feral hogs on forest dynamics and processes in the southeastern US. Forest Ecology and Management 258, 546–553 (2009).
Wirthner, S. et al. Do changes in soil properties after rooting by wild boars (Sus scrofa) affect understory vegetation in Swiss hardwood forests? Can. J. Forest Res. 42, 585–592 (2012).
Bruinderink, G. G. & Hazebroek, E. Wild boar (Sus scrofa scrofa L.) rooting and forest regeneration on podzolic soils in the Netherlands. Forest Ecol. Manag. 88, 71–80 (1996).
Doan, T. T., Jusselme, D. M., Lata, J. C., Nguyen, B. V. & Jouquet, P. The earthworm species Metaphire posthuma modulates the effect of organic amendments (compost vs. vermicompost from buffalo manure) on soil microbial properties. a laboratory experiment. Eur. J. Soil Biol. 59, 15–21 (2013).
Kaller, M. D. & Kelso, W. E. Swine activity alters invertebrate and microbial communities in a coastal plain watershed. Am. Midl. Nat. 156, 163–177 (2006).
Yang, S. H. et al. Characterization of microbial community in the leachate associated with the decomposition of entombed pigs. J. Microbiol. Biotechn. 22, 1330–1335 (2012).
Parkes, J. P., Easdale, T. A., Williamson, W. M. & Forsyth, D. M. Causes and consequences of ground disturbance by feral pigs (Sus scrofa) in a lowland New Zealand conifer-angiosperm forest. New Zeal. J. Ecol. 39, 34–42 (2015).
Wirthner, S., Frey, B., Busse, M. D., Schutz, M. & Risch, A. C. Effects of wild boar (Sus scrofa L.) rooting on the bacterial community structure in mixed-hardwood forest soils in Switzerland. Eur. J. Soil Biol. 47, 296–302 (2011).
Fujino, C. et al. Effect of different organic amendments on the resistance and resilience of the organic matter decomposing ability of soil and the role of aggregated soil structure. Soil Sci. Plant Nutr. 54, 534–542 (2008).
Allison, S. D. & Martiny, J. B. Resistance, resilience, and redundancy in microbial communities. P. Natl. Acad. Sci. USA 105, 11512–11519 (2008).
Buscardo, E. et al. Of mammals and bacteria in a rainforest: temporal dynamics of soil bacteria in response to simulated N pulse from mammalian urine. Funct. Ecol. 32, 773–784 (2018).
Zogg, G. P. et al. Compositional and functional shifts in microbial communities due to soil warming. Soil Sci. Soc. Am. J. 61, 475–481 (1997).
Becker, J. M., Parkin, T., Nakatsu, C. H., Wilbur, J. D. & Konopka, A. Bacterial activity, community structure, and centimeter-scale spatial heterogeneity in contaminated soil. Microb. Ecol. 51, 220–231 (2006).
Nottingham, A. T. et al. Microbes follow Humboldt: temperature drives plant and soil microbial diversity patterns from the Amazon to the Andes. Ecology 99, 2455–2466 (2018).
Bell, T. et al. Microbial biodiversity and ecosystem functioning under controlled conditions and in the wild in Biodiversity, ecosystem functioning, and human wellbeing: an ecological and economic perspective (eds Naeem, S., Bunker, D. E., Hector, A., Loreau, M. & Perrings, C.) 121–133 (Oxford University Press, 2009).
NRCS. S survey staff, Natural Resources Conservation Services, United States Department of Agriculture. Web Soil Survey, http://websoilsurvey.nrcs.usda.gov/ (2018).
Giambelluca, T. W. et al. Online Rainfall Atlas of Hawaii. Bull. Am. Meterolo. Soc. 94, 313–316 (2013).
Wagner, W. L., Herbst, D. R. & Sohmer, S. H. Manual of the flowering plants of Hawaii. (University of Hawaii Press, 1999).
Scheffler, P. Y., Pratt, L. W., Foote, D. & Magnacca, K. N. A preliminary study of effects of feral pig density on native Hawaiian montane rainforest vegetation. Technical Report at, https://scholarspace.manoa.hawaii.edu/handle/10125/34420 (2012).
Illumina. 16S metagenomic sequencinc library preparation: preparing 16S ribosomal RNA gene amplicons for the Illumina Miseq System. Guide at, https://support.illumina.com/downloads/16s_metagenomic_sequencing_library_preparation.html (2013).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1, https://doi.org/10.1093/nar/gks808 (2013).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Langille, M. G. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013).
RStudio Team. RStudio: integrated development for R. Software at, http://www.rstudio.com (2015).
Oksanen, J. et al. Package ‘vegan’. Software at, http://cran.r-project.org/web/packages/vegan/vegan.pdf (2013).
Wehr, N.H. Responses of soil invertebrate and bacterial communities to the removal of nonnative feral pigs (Sus scrofa) from a Hawaiian tropical montane wet forest. (University of Hawai'i at Manoa, 2018).
Acknowledgements
We would like to acknowledge the following people for their valuable contributions to this project: Kainana Francisco for logistical support; Renee Corpuz, Dr. Jenifer Saito, and Anne Veillet for laboratory assistance; and Nanea Babila, Nathanael Friday, and Taylor Tomita for fieldwork assistance. In addition, we thank Dr. Steven Hess and Dr. Noa Lincoln for their helpful manuscript reviews. Finally, we would like to thank the National Park Service and the State of Hawai’i Department of Land and Natural Resources, Division of Forestry and Wildlife for access to research sites. This work was supported by the Watson T. Yoshimoto Fellowship distributed by the Ecology, Evolution, and Conservation Biology Program at the University of Hawai’i at Mānoa, the University of Hawai’i at Mānoa’s Graduate Student Organization Grants and Awards Program, the College of Tropical Agriculture and Human Resources, University of Hawai’i at Mānoa via the USDANIFA Hatch (HAW01127H) and McIntyre Stennis (HAW01123M) Programs, the Department of Defense Strategic Environmental Research and Development Program (RC-2433), and the U.S. Forest Service. Additionally, this work was supported by the completion of the thesis written by N.H.W.55.
Author information
Authors and Affiliations
Contributions
C.M.L., C.P.G. and K.M.K. were involved in study design. N.H.W. collected samples, which were extracted and sequenced by N.H.W. and K.M.K. QIIME2 script production and data analysis were conducted by N.H.W. under the guidance of N.H.N., N.H.W. wrote the first draft of the manuscript. All authors discussed the results and commented on or edited the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wehr, N.H., Kinney, K.M., Nguyen, N.H. et al. Changes in soil bacterial community diversity following the removal of invasive feral pigs from a Hawaiian tropical montane wet forest. Sci Rep 9, 14681 (2019). https://doi.org/10.1038/s41598-019-48922-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48922-7
This article is cited by
-
Inconsistent recovery of nitrogen cycling after feral ungulate removal across three tropical island ecosystems
Biogeochemistry (2024)
-
Wild boar rooting impacts soil function differently in different plant community types
Biological Invasions (2023)
-
Historical range expansion and biological changes of Sus scrofa corresponding to domestication and feralization
Mammal Research (2021)
-
Relationships between soil macroinvertebrates and nonnative feral pigs (Sus scrofa) in Hawaiian tropical montane wet forests
Biological Invasions (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
