
Abstract
Liquid biopsies of circulating tumor DNA (ctDNA) have recently been used as a non-invasive diagnostic tool for detecting tumor-specific mutations. We present a study of ctDNA liquid biopsies in gynecological cancer using an ultrasensitive next-generation sequencing-based method for ctDNA detection named CAncer Personalized Profiling by deep Sequencing (CAPP-Seq). We performed CAPP-Seq with plasma-ctDNA obtained from 16 patients with gynecological cancer. In all cases, at least one non-synonymous somatic mutation was detected in the ctDNA. In the pre-treatment ctDNA, 4 of 16, 4/16, 5/16, 2/16, 2/16, and 2/16 patients had TP53, KRAS, APC, PIK3CA, BRCA1, and EGFR mutations, respectively. MET gene copy-number gains were detected in the ctDNA of 2 of 16 patients, and FISH analysis of the paired tumor samples confirmed these results. In 2 neoadjuvant chemotherapy-treated ovarian cancer patients, the changes in gene mutation patterns were associated with the treatment response. These findings suggest that CAPP-Seq-based liquid biopsies can be used for the genetic characterization of independent gynecological cancers with high frequency, and might be clinically useful for non-invasive tumor genotyping and therapeutic response monitoring.
Similar content being viewed by others
Introduction
Recent studies have shown that genomic alterations in solid cancers can be characterized by sequencing circulating tumor DNA (ctDNA)1,2,3. This technique is effectively a form of “liquid biopsy”, and such examinations are more widely available and easier to process than standard tumor biopsies4,5. DNA fragments are released into the bloodstream from apoptotic or necrotic cells6. In patients with solid tumors, ctDNA is also released via necrosis, autophagy, apoptosis, and other physiological events induced by microenvironmental stress as well as the effects of treatment7. Recent improvements in polymerase chain reaction (PCR)-based assays for analyzing blood samples for ctDNA have provided rapid, cost-effective, and non-invasive alternatives to tumor biopsies. These methods provide information about molecular alterations due to point mutations, including tumor-specific mutations, and have been used as diagnostic, prognostic, and therapeutic decision-making tools, especially in lung cancer (to detect epidermal growth factor receptor [EGFR] and Kirsten rat sarcoma viral oncogene homolog [KRAS] mutations) and colorectal cancer (to detect RAS mutations)8,9,10. Furthermore, the analysis of ctDNA liquid biopsy samples, whose results might not be influenced by heterogeneity, could aid the design of effective treatment strategies11. However, most early-advanced stage solid tumors exhibit very low levels of ctDNA, which makes difficult to detect genomic alterations. Although extensive research into the genetic profiling of gynecological cancers has been conducted using tumor tissue-derived DNA sequencing12,13,14, the ctDNA liquid biopsy technique has not been commonly used in the gynecological oncology field.
To improve ctDNA detection, an ultrasensitive next-generation sequencing-based (NGS-based) method for ctDNA detection named CAncer Personalized Profiling by deep Sequencing (CAPP-Seq) was recently established15,16. CAPP-Seq is the first NGS-based ctDNA analysis method that achieves both an ultralow detection limit and broad patient coverage at a reasonable cost and low ctDNA input level, and hence, it allows the quantitation of ctDNA from early stage tumors. CAPP-Seq achieves similar sensitivity at hotspot alleles as digital PCR or amplicon-based approaches, but is able to simultaneously interrogate thousands of additional genomic positions without its sensitivity or specificity being affected. This ultrasensitive technique can detect ctDNA in patients with early and advanced stages of various human malignancies, including lung cancer, lymphoma, and leiomyosarcoma16,17,18. In the gynecological oncology field, there have been few reports about the comprehensive gene mutation analysis of liquid biopsy samples19,20,21. Thus, we first conducted a study of multiple gene alteration analysis of ctDNA using CAPP-Seq in patients with gynecological cancer, and compared the findings with those obtained using tumor DNA sequencing.
Results
Clinical characteristics of the study cohort
Of the 16 patients included in this study, 2 had metastases from colorectal cancer to the ovary (COL), 4 had primary ovarian cancer (OVA), 4 had cervical cancer (CER), 5 had endometrial cancer (END), and one had uterine leiomyosarcoma (LMS). Information regarding tumor stage, treatment, and histopathological features are summarized in Table 1. Of the 16 patients, 6, 2, 4, and 3 patients were diagnosed with stage I, stage II, stage III, and stage IV disease, respectively. Pre-treatment plasma samples were available for 14 of the 16 patients who underwent surgical resection or CCRT (50 Gy radiotherapy combined with 6 cycles of weekly 40 mg/m2 cisplatin). Paired pre-neoadjuvant chemotherapy (NAC) and post-NAC (pre-surgery) plasma samples were available for 2 of the 16 patients who underwent interval debulking surgery after NAC. Three cycles of paclitaxel (175 mg/m2) and carboplatin (area under the curve: 5.0, according to Calvert’s formula) were administered every 3–4 weeks as NAC.
Performance of CAPP-Seq of ctDNA from patients with gynecological cancer
The ctDNA extracted from the pre-treatment plasma samples was subjected to CAPP-Seq using a gene-sequencing panel containing 197 target genes. At least one non-synonymous somatic mutation (median: 3, range: 1–13 mutations/sample) was detected in the samples from each of the 16 patients (16/16; 100%). The number of non-synonymous somatic mutations tended to be higher in the stage IV group, although the difference was not significant (Fig. 1A). However, there were highly mutated cases in both the advanced stage and early stage groups (Fig. 1A). The number of mutations varied among the different types of cancer, with endometrial cancer having the lowest number of mutations (Fig. 1B). The highest mutant allele fraction tended to be greater in the stage IV group, although the differences between the stage IV group and the other groups were not statistically significant (Fig. 1C). There were no significant differences in the highest mutant allele fraction among the various cancer types (Fig. 1D).

CAPP-Seq-based ctDNA analysis in patients with gynecological cancer. (A) The numbers of non-synonymous somatic mutations detected in the ctDNA of cancer patients according to disease stage. (B) The numbers of non-synonymous somatic mutations detected in the ctDNA of cancer patients according to cancer type. (C) Mutant allele fractions of ctDNA according to disease stage. (D) Mutant allele fractions of ctDNA according to cancer type. The median values for each group are represented by black bars. For patients in whom multiple alterations were detected, the highest value is shown.
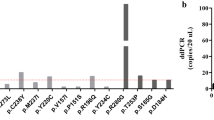
We identified 68 non-synonymous somatic mutations across 44 genes in the pre-treatment plasma ctDNA of the 16 patients (Fig. 2). In total, 4 (25%), 4 (25%), 5 (31%), 2 (13%), 2 (13%), and 2 (13%) patients had tumor protein 53 (TP53), KRAS, adenomatous polyposis coli (APC), phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA), breast cancer gene 1 (BRCA1), and EGFR mutations, respectively. Of the 68 non-synonymous somatic mutations detected in the plasma ctDNA, 13 were also observed in the paired tumor DNA (analyzed by QIAseq using a gene sequencing panel containing 275 target genes). Concordance between the mutations detected in the plasma and tumor DNA was evident in four cancer types; i.e., in 2 of 2 (100%) patients with metastasis from colorectal cancer to the ovary, 3 of 4 (75%) patients with ovarian cancer, 2 of 4 (50%) patients with cervical cancer, and 1 of 5 (20%) patients with endometrial cancer. The plasma/tumor-matched mutations included 4 TP53 mutations (H193Y, Y205C, Y234N, and R248W), 2 KRAS mutations (G12D and G13D), 2 APC mutations (R216* and E1306*), one PIK3CA mutation (E545K), 2 EGFR mutations (A611T and R831H), and one nuclear factor (erythroid-derived 2)-like 2 (NFE2L2) mutation (R34G).

Summary of ctDNA gene alterations identified by CAPP-Seq. *Gene mutations identified in both tumor and plasma samples.
Copy-number amplification in the ctDNA extracted from the plasma samples
We also detected somatic copy-number amplification in two genes, MET and EGFR, in the plasma ctDNA using CAPP-Seq (Fig. 2). MET copy-number amplifications were observed in 2 patients (13%), and EGFR copy-number amplifications were observed in 5 patients (31%). To investigate the validity of the MET copy-number amplification detected by CAPP-Seq, the paired tumor FFPE specimens were subjected to FISH analysis of MET amplification (Fig. 3). In the 2 patients that displayed MET amplification during CAPP-Seq (COL1 and CER3), 48.3% (Fig. 3A,C) and 41.7% (Fig. 3C) of the FFPE specimen cells contained ≥4 copies of the MET gene, respectively. While in 2 patients that did not demonstrate MET amplification during CAPP-Seq (OVA4 and END1), 8.4% of FFPE specimen cells contained ≥4 copies of the MET gene in OVA4 (Fig. 3B,D), and 11.7% of FFPE specimen cells were in END1 (Fig. 3D).

FISH analysis of the MET gene. (A) Image of a FISH-positive specimen (COL1). (B) Image of a FISH-negative specimen (OVA4). (C) Number of MET signals/cell in FISH-positive specimens (COL1 and CER3). (D) Number of MET signals/cell in FISH-negative specimens (OVA4 and END1).
Plasma ctDNA monitoring for assessing the response to NAC
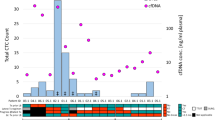
Our results demonstrated the possibility of using CAPP-Seq for ctDNA to assess treatment responses in ovarian cancer patients treated with NAC (Fig. 4). In 2 ovarian cancer patients, OVA2 and OVA3, ctDNA was isolated from both their pre-NAC and post-NAC plasma samples, and CAPP-Seq was performed to monitor the changes in the gene mutation patterns seen after NAC. In the NAC-sensitive case OVA2 (Fig. 4A), the sum of the longest diameters (SLD) of the tumor decreased from 21 to 9 cm, and the cancer antigen 125 (CA125) level decreased from 8275 to 52.9 U/mL. The maximum standardized uptake value (SUVmax) on 18F-fluoro-2-deoxy-D-glucose positron emission tomography combined with computed tomography (FDG-PET/CT), which is a parameter of tumor aggressiveness, also decreased from 12.4 to 2.96. We detected one TP53 mutation (R248W; mutation level: 4.42 mutant copies per mL) in the patient’s pre-NAC ctDNA via CAPP-Seq, while no TP53 (R248W) mutations were detected in their post-NAC ctDNA. In the analysis of the post-NAC FFPE tissue sample from OVA2, we detected the same TP53 mutation (R248W) as was detected in the patient’s pre-NAC ctDNA. In the NAC-resistant case OVA3 (Fig. 4B), the SLD of the tumor increased from 17 to 19 cm, and the patient’s CA19-9 level increased from 6342 to 8690 U/mL. We detected one KRAS mutation (G12D; mutation level: 3.04 mutant copies per mL) in the patient’s pre-NAC ctDNA via CAPP-Seq, while their post-NAC ctDNA exhibited an increased KRAS (G12D) mutation level (14.6 mutant copies per mL) and 3 novel TP53 mutations (Y234N, V143A, and H193A). In the analysis of the post-NAC FFPE tissue sample from OVA3, we detected the same KRAS (G12D) mutation and one of the TP53 (Y234N) mutations that were detected in the patient’s post-NAC ctDNA.

Changes in gene mutation profiles of ctDNA and clinical courses in NAC-treated ovarian cancer cases. Comparison of gene mutations in pre- and post-NAC ctDNA and the clinical courses of (A) an NAC-sensitive ovarian cancer case (OVA2) and (B) an NAC-resistance ovarian cancer case (OVA3).
Discussion
To the best of our knowledge, the current study is the first to demonstrate the utility of analyzing comprehensive gene alterations of liquid biopsy-derived ctDNA using CAPP-Seq in patients with gynecological cancer. Using CAPP-Seq, we detected at least one non-synonymous somatic mutation in the plasma ctDNA of all 16 gynecological cancer patients, and 8 of 16 patients had mutations in their plasma that matched mutations found in their paired tumor samples. These findings suggest that CAPP-Seq-based ctDNA profiling of gynecological malignancies is useful.
The use of targeted hybrid capture with high-throughput sequencing and a specialized bioinformatics workflow technique for plasma ctDNA allows the highly sensitive, non-invasive, and low-cost ctDNA-detection15,16. CAPP-Seq has been demonstrated to be clinically useful for improving prognostication in lymphoma17 and for studying the heterogeneity of resistance mechanisms in lung cancer patients22. Plasma genotyping of EGFR-sensitizing mutations has already been approved by the US Food and Drug Administration as a technique for selecting feasible drugs10,23. In our study, we determined the gene mutation profile of ctDNA using CAPP-Seq, not only in gynecological cancer, but also in metastasis from colorectal cancer to the ovary, which exhibited the well-known genetic signature of colorectal cancer (KRAS, APC, and TP53 mutations and a MET copy-number gain). Therefore, CAPP-Seq might be applicable to a broad range of clinical cases and have the potential to accelerate the personalized detection, treatment, and monitoring of many kinds of cancer.
In the gynecological oncology field, there have only been a few reports about gene mutation analysis using ctDNA. We revealed that CAPP-Seq-based examinations have the potential to facilitate the characterization of the genetic profiles of gynecological cancer. All 16 of the analyzed plasma samples had at least one detectable non-synonymous somatic mutation according to the ultrasensitive NGS-based method employed in this study. FFPE tumor samples were also examined, and the same mutation was detected in both the tumor and plasma samples in 8 patients. These results suggest that CAPP-Seq is feasible as an ultrasensitive method for performing comprehensive gene mutation analysis of gynecological cancer, as has been reported for lung cancer, lymphoma, and leiomyosarcoma16,17,18. Chabon et al. used CAPP-Seq to examine the intra-patient tumor heterogeneity that arose during treatment for non-small cell lung cancer22. Among 41 patients who exhibited T790M mutations at enrollment after receiving EGFR-targeted therapy, additional putative resistance mutations were detected in the pretreatment plasma ctDNA of 19 patients (46%), including MET copy-number gains, the target gene alteration for the MET inhibitor crizotinib, which we also detected in 2 patients using both CAPP-Seq of ctDNA and FISH analysis of tumor samples (COL1 and CER3). Przybyl et al. reported that ctDNA monitoring using CAPP-Seq was clinically useful in 7 cases of leiomyosarcoma, including 3 cases of uterine leiomyosarcoma18. In the latter study, gene mutations were detected in the ctDNA of 6 of 7 leiomyosarcoma patients using CAPP-Seq, and it was suggested that CAPP-Seq has potential as a method for assessing tumor heterogeneity. In our case LMS1, although we could not detect any gene mutations in 3 FFPE tumor specimens, we detected non-synonymous somatic mutations in the KRAS and receptor tyrosine-protein kinase erbB-2 (ERBB2) genes, both of which are known to be important actionable genes, in the analysis of the patient’s ctDNA. Further studies are needed to confirm the usefulness of CAPP-Seq for assessing tumor heterogeneity in gynecological cancer.
Ovarian cancer is often diagnosed at an advanced stage, and little progress has been made in the existing chemotherapies for the disease24. Although there are several new therapeutic concepts that can be used to treat some cases of ovarian cancer, including targeted therapy involving the vascular endothelial growth factor inhibitor bevacizumab or the poly-(ADP-ribose) polymerase inhibitor olaparib25,26, ovarian cancer still causes the majority of cancer-related deaths from gynecological cancer27. Thus, a new treatment strategy for ovarian cancer, including novel approaches for early diagnosis, tumor monitoring, and/or detecting novel molecular targets, is needed. There have only been a few studies about the comprehensive gene mutation analysis of ctDNA in the gynecological oncology field, and they involved small numbers of patients19,20,21. Phallen et al.19 used targeted error correction sequencing, which allows direct ultrasensitive evaluations of 58 cancer-related genetic alterations, to analyze ctDNA from four types of cancer (colorectal, lung, ovarian, and breast cancer). In their study, of the 42 ovarian cancer patients included, 27 (64%) had at least one detectable somatic mutation in their plasma ctDNA (range: 1–3 mutations/sample), and about 50% of the ovarian cancer patients had TP53 mutations. Cohen et al.20 used CancerSEEK, a combined assay for genetic alterations in 16 cancer-related genes and 41 protein biomarkers, to analyze eight types of cancer (colorectal, lung, ovarian, breast, liver, stomach, pancreatic, and esophageal cancer). Although the 54 ovarian cancer patients included in the latter study had detectable somatic mutations in their plasma ctDNA, only one somatic mutation was detected in each patient, and 46 (85%) patients had TP53 mutations. Arend et al.21 used a basic NGS method (the Ion AmpliSeq cancer hotspot panel v2) for ultra-deep targeted sequencing of ctDNA from 14 patients with advanced ovarian cancer (stage III–IV). Although they detected 12 variants in matched tumor and plasma ctDNA samples, it was considered likely that all 12 of these variants were germline mutations. In our study, all of the ovarian cancer patients (4/4; 100%) had at least one detectable non-synonymous somatic mutation, and 3 of the 4 patients had identical non-synonymous somatic mutations in their matched tumor and plasma ctDNA samples. Indeed, our results demonstrate the possibility of performing ctDNA liquid biopsy examinations using CAPP-Seq for monitoring treatment responses, including “tumor evolution28”; i.e., evolutionary changes in tumor heterogeneity, in NAC-treated patients with advanced ovarian cancer (OVA2 and OVA3). In our NAC-sensitive case OVA2 (Fig. 4A), CAPP-Seq detected a reduction in the TP53 (R248W) mutation level after NAC treatment. In contrast, in the NAC-resistant case OVA3 (Fig. 4B), we detected an increase in the KRAS (G12D) mutation level and the appearance of 3 novel TP53 mutations (Y234N, V143A, and H193A) after NAC treatment. Thus, CAPP-Seq might be a useful tool for non-invasive tumor genotyping as well as for therapeutic response monitoring, detecting evolutional changes in tumor heterogeneity.
The information about ctDNA liquid biopsies of uterine cancer (cervical and endometrial cancers) is even more limited than that about ovarian cancer29,30. There have not been any reports about the comprehensive gene mutation analysis of ctDNA in uterine cancer, and the current study is the first to characterize the ctDNA genetic profile of uterine cancer by CAPP-Seq. Chung et al. assessed the frequency of 2 major PIK3CA mutations in uterine cervical cancer, E542K and E545K, using droplet digital PCR of plasma ctDNA, and the detection of PIK3CA mutations in plasma was associated with reduced disease-free survival and overall survival29. Bolivar et al. used a custom-designed Accel-Amplicon NGS panel covering hotspots in the phosphatase and tensin homolog (PTEN), catenin beta-1 (CTNNB1), KRAS, and PIK3CA genes to analyze ctDNA from 48 endometrial cancer patients30. At least one mutation was detected in the ctDNA of 17 (35%) patients, and the same mutation was detected in both tumor and plasma samples in 15 of 45 patients (33%), which demonstrated that mutations in endometrial cancer-related genes can be identified in plasma ctDNA. We detected at least one non-synonymous somatic mutation in all of the uterine cancer patients in this study (CER and END), and identical non-synonymous somatic mutations were detected in matched tumor and plasma ctDNA samples in 3 of the 9 patients (CER2, CER4, and END5). Indeed, we also detected a MET copy-number gain in one patient (CER3) and EGFR copy-number gains in 2 patients (CER2 and CER3), both of which could be targets for novel molecular treatments for uterine cancer.
Interestingly, our study showed that there might be populations of highly mutated tumors in both ovarian and uterine cancer, which could be detected by CAPP-Seq (e.g., OVA1, CER2, and END5). Rizvi et al. reported that the tumor mutation burden (TMB), as determined by targeted NGS, might be associated with the response to immunotherapy in patients with lung cancer31. Gandara et al. demonstrated that the TMB according to blood tests (blood-TMB; bTMB) might be associated with the clinical benefit from immune checkpoint inhibitor therapy in patients with lung cancer32. In addition, it was reported that CAPP-Seq has potential as a technique for measuring the bTMB among early-advanced stage cancer and for monitoring ctDNA during treatment15,16. Further study is necessary to determine the utility of performing ctDNA liquid biopsies using CAPP-Seq for evaluating the bTMB in the gynecological field.
Collectively, the present study has demonstrated the utility of performing ctDNA liquid biopsy examinations, which are non-invasive and easy to perform repeatedly, using the novel ultrasensitive CAPP-Seq method in patients with gynecological cancer. Genetic characterization of gynecological cancers based on CAPP-Seq might aid the development of novel personalized treatment strategies. There are some limitations to our study. We did not utilize the tumor DNA for CAPP-Seq in the same gene-sequencing panel. CAPP-Seq is currently specialized for plasma ctDNA, and it therefore remains a future challenge to expand its application to formalin-fixed paraffin-embedded tumor tissue. Further experiments using the tumor DNA are needed to explore the ability/sensitivity to detect the tumor mutations by ctDNA. Another limitation is that we conducted the retrospective study cohort with a relatively small number of patients. Further studies should be in a large number of patients conducted to clarify the clinical usefulness of CAPP-Seq-based ctDNA profiling.
Material and Methods
Ethical approval and informed consent
The present study was approved by the ethics committees of Wakayama Medical University (Authorization Number: 2025) and Kindai University Faculty of Medicine (Authorization Number: 29-066). All of the patients in this study provided written informed consent for the use of their plasma and tissue samples. All methods were performed in accordance with the Declaration of Helsinki specified by Wakayama Medical University and Kindai University Faculty of Medicine.
Patients and samples
A total of 16 patients with gynecological cancer who underwent surgical resection, chemotherapy, or concurrent chemoradiotherapy (CCRT) at Wakayama Medical University Hospital between May 2017 and January 2018 were included in this study. The tumor staging was carried out according to the International Federation of Gynecology and Obstetrics (FIGO) classification, and tumor grading was performed according to the World Health Organization (WHO) classification. All patients were pathologically diagnosed using surgical resection or biopsy samples. Histological diagnosis was determined on the basis of standard H&E-stained sections by two or more experienced senior pathologists.
Circulating tumor DNA extraction and sequencing
CAPP-Seq was performed with ctDNA isolated from plasma samples as previously reported16. In brief, we used a maximum of 50 ng of DNA for the CAPP-Seq ctDNA analyses, which were performed using the AVENIO ctDNA surveillance kit for 197 genes (Roche Diagnostics, Indianapolis, IN), according to the manufacturer’s instructions. The AVENIO ctDNA Surveillance Kit contains major mutated genes in gynecological cancer (TP53, BRCA1, BRCA2, BRAF, PIK3CA, KRAS, NRAS, APC, EGFR, MET, ERBB2, CTNNB1, etc.). The purified libraries were pooled and sequenced on an Illumina NextSeq. 500 (Illumina, San Diego, CA) using a 300-cycle high-output kit. Variants were called with the AVENIO ctDNA analysis software (Roche Diagnostics), which includes bioinformatics methods from CAPP-Seq15 and integrated digital error suppression16. Germline mutations were excluded based on the Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB) and the Exome Aggregation Consortium database33.
Tumor DNA extraction and sequencing
We analyzed 40 ng of DNA with the QIAseq Human Comprehensive Cancer Panel for 275 genes (Qiagen, Valencia, CA). The library preparation was performed according to the manufacturer’s instructions34. The purified libraries were pooled and then sequenced with a NextSeq. 500 instrument (Illumina). Reads were aligned with the hg19 human reference genome, and variant detection was performed according to the manufacturer’s pipeline. Germline mutations were excluded using the Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB) and the Exome Aggregation Consortium database33.
Fluorescence in situ hybridization analysis (FISH)
Amplification of the MET gene from tumor tissue was examined via FISH analysis. Four-micrometer FFPE specimens were hybridized with the MET (TexRed)/CEN7q (FITC) dual-color FISH probe (GSP Laboratory, LCI Medience Corporation, Chiba, Japan). The number of fluorescent signals was counted independently by two investigators. Positivity for MET gene amplification was defined as follows: over 30% of cells exhibiting ≥4 copies of the MET gene. These examinations were outsourced to LSI Medience Corporation (Tokyo, Japan).
Statistical methods
The statistical analyses were performed using the Mann-Whitney U test. P-values of <0.05 were considered significant.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Blair, B. G., Bardelli, A. & Park, B. H. Somatic alterations as the basis for resistance to targeted therapies. J Pathol 232, 244–254 (2014).
Schwarzenbach, H., Hoon, D. S. & Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 11, 426–437 (2011).
McDermott, U., Downing, J. R. & Stratton, M. R. Genomics and the continuum of cancer care. N Engl J Med 364, 340–350 (2011).
Misale, S. et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486, 532–536 (2012).
Murtaza, M. et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497, 108–112 (2013).
Stroun, M., Lyautey, J., Lederrey, C., Olson-Sand, A. & Anker, P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta 313, 139–142 (2001).
Delgado, P. O. et al. Characterization of cell-free circulating DNA in plasma in patients with prostate cancer. Tumour Biol 34, 983–986 (2013).
Peeters, M. et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res 19, 1902–1912 (2013).
Qiu, M. et al. Circulating tumor DNA is effective for the detection of EGFR mutation in non-small cell lung cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev 24, 206–212 (2015).
Oxnard, G. R. et al. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J Clin Oncol 34, 3375–3382 (2016).
Garcia-Murillas, I. et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med 7, 302–133 (2015).
Cancer Genome Arlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011).
Cancer Genome Arlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013).
Cancer Genome Arlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 543, 378–384 (2017).
Newman, A. M. et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 20, 548–554 (2014).
Newman, A. M. et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 34, 547–555 (2016).
Scherer, F. et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med 8, 364–155 (2016).
Przybyl, J. et al. Combination Approach for Detecting Different Types of Alterations in Circulating Tumor DNA in Leiomyosarcoma. Clin Cancer Res 24, 2688–2699 (2018).
Phallen, J. et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med 9, eaan2415 (2017).
Cohen, J. D. et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926–930 (2018).
Arend, R. C. et al. Molecular Response to Neoadjuvant Chemotherapy in High-Grade Serous Ovarian Carcinoma. Mol Cancer Res 16, 813–824 (2018).
Chabon, J. J. et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun 7, 11815 (2016).
Wan, R. et al. Comprehensive Analysis of the Discordance of EGFR Mutation Status between Tumor Tissues and Matched Circulating Tumor DNA in Advanced Non-Small Cell Lung Cancer. J Thorac Oncol 12, 1376–1387 (2017).
Du Bois, A. & Pfisterer, J. Future options for first-line therapy of advanced ovarian cancer. Int J Gynecol Cancer 15(Suppl 1), 42–50 (2005).
Burger, R. A. et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med 365, 2473–2483 (2011).
Ledermann, J. et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 15, 852–861 (2014).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2015, CA. Cancer J Clin 65, 5–29 (2015).
McGranahan, N. & Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 27, 15–26 (2015).
Chung, T. K. H. et al. Liquid biopsy of PIK3CA mutations in cervical cancer in Hong Kong Chinese women. Gynecol Oncol 146, 334–339 (2017).
Bolivar, A. M. et al. Targeted next-generation sequencing of endometrial cancer and matched circulating tumor DNA: identification of plasma-based, tumor-associated mutations in early stage patients. Mod Pathol 32, 405–414 (2019).
Rizvi, H. et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J Clin Oncol 36, 633–641 (2018).
Gandara, D. R. et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med 24, 1441–1448 (2018).
Narahara, M. et al. Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS One 9, e100924 (2014).
Xu, C., Nezami Ranjbar, M. R., Wu, Z., DiCarlo, J. & Wang, Y. Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller. BMC Genomics 18, 5 (2017).
Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research (Grant No. JP18K16776) from the Japan Society for the Promotion of Science. No other specific grants or funding were received.
Author information
Authors and Affiliations
Contributions
N.I. performed the experiments, interpreted the data, and wrote the paper. K.S. and T.N. analyzed the circulating tumor DNA sequencing and interpreted the data. K.S., T.Y., H.M. and S.T. analyzed the tumor DNA sequencing and interpreted the data. K.S. contributed reagents, materials, and analytical tools and supervised the entire project. K.N. and K.I. designed the research, interpreted the data, and wrote the paper and takes full responsibility for the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Iwahashi, N., Sakai, K., Noguchi, T. et al. Liquid biopsy-based comprehensive gene mutation profiling for gynecological cancer using CAncer Personalized Profiling by deep Sequencing. Sci Rep 9, 10426 (2019). https://doi.org/10.1038/s41598-019-47030-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47030-w
This article is cited by
-
Potential clinical utility of liquid biopsies in ovarian cancer
Molecular Cancer (2022)
-
Clinical applications and utility of cell-free DNA-based liquid biopsy analyses in cervical cancer and its precursor lesions
British Journal of Cancer (2022)
-
Promising Blood-Based Biomarkers for Melanoma: Recent Progress of Liquid Biopsy and Its Future Perspectives
Current Treatment Options in Oncology (2022)
-
Prediction of the treatment response in ovarian cancer: a ctDNA approach
Journal of Ovarian Research (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
