
Abstract
Wildlife is a reservoir of emerging infectious diseases of humans and domestic animals. Marmota himalayana mainly resides 2800–4000 m above sea level in the Qinghai-Tibetan Plateau, and is the primary animal reservoir of plague pathogen Yersinia pestis. Recently we isolated a new species, Escherichia marmotae from the faeces of M. himalayana. In this study we characterised E. marmotae by genomic analysis and in vitro virulence testing to determine its potential as a human pathogen. We sequenced the genomes of the seven E. marmotae strains and found that they contained a plasmid that carried a Shigella-like type III secretion system (T3SS) and their effectors, and shared the same O antigen gene cluster as Shigella dysenterae 8 and E. coli O38. We also showed that E. marmotae was invasive to HEp-2 cells although it was much less invasive than Shigella. Thus E. marmotae is likely to be an invasive pathogen. However, E. marmotae has a truncated IpaA invasin, and lacks the environmental response regulator VirF and the IcsA-actin based intracellular motility, rendering it far less invasive in comparison to Shigella. E. marmotae also carried a diverse set of virulence factors in addition to the T3SS, including an IS1414 encoded enterotoxin gene astA with 37 copies, E. coli virulence genes lifA/efa, cif, and epeA, and the sfp gene cluster, Yersinia T3SS effector yopJ, one Type II secretion system and two Type VI secretion systems. Therefore, E. marmotae is a potential invasive pathogen.
Similar content being viewed by others

Introduction
Wildlife is a reservoir of emerging infectious diseases of humans and domestic animals1. These include both viral and bacterial pathogens. Wild waterfowl is the natural reservoir of all known subtypes of the influenza A virus, some of which contributes to the genesis of new subtypes to cause pandemics in humans2. Lyme disease caused by the bacterium Borrelia burgdorferi emerged from human contact with wild deer population in North America which carries the tick borne disease3. Proactive surveillance of wildlife for novel pathogens will help us to predict, prevent and manage emerging disease threats to humans.
Marmots are large terrestrial rodents widespread across much of northern Eurasia and North America4. Marmota himalayana mainly resides 2800–4000 m above sea level in the Qinghai-Tibetan Plateau, and is the primary animal reservoir of Yersinia pestis, the causative agent of bubonic plague5. Recently, we identified a number of novel bacterial species in wild Marmots6,7,8,9. With the expansion of human activity in the Qinghai-Tibetan Plateau, the chances of human–M. himalayana interaction have increased, which may allow transmission of pathogens from M. himalayana to humans. We recently analysed E. coli from faecal samples of M. himalayana and found multiple pathogenic types of E. coli present in these faecal samples10. We also isolated a new species, Escherichia marmotae, from the faeces of M. himalayana11.
In this study, we characterised E. marmotae by genomic analysis and in vitro virulence testing and found that it is a potential invasive pathogen with Shigella-like invasion genes. Shigella is a human only invasive pathogen and can invade intestinal epithelial cells, M cells, macrophages and interact with the intestinal mucosa, leading to bacillary dysentery12 Shigella carries an invasive plasmid, generally referred to as pINV which is also shared by enteroinvasive E. coli (EIEC)13, which encodes a type III secretion system (T3SS) for tissue invasion and other factors for its intracellular lifestyle14,15. For Shigella, the T3SS translocates a set of approximately 25 proteins from the bacterial cytoplasm directly into the eukaryotic host cell, where these “effector” proteins interfere with various host cell processes14,15. We found that E. marmotae from wild rodents carried a Shigella-like T3SS and associated effectors on a plasmid. The E. marmotae genome also contained a range of other virulence genes. Our results suggested that E. marmotae is a potential invasive pathogen.
Results
Genome sequencing of E. marmotae
The seven E. marmotae strains, HT073016, HT080709, HT080711, HT072503, HT073105, HT080118 and HT080401, isolated from the faeces of seven healthy M. himalayana in the Qinghai-Tibet plateau in China11, were sequenced using the Illumina Hi-Seq platform as draft genomes and the E. marmotae type strain HT073016 was further completely sequenced by Pacific Biosciences SMRT sequencing. The genome of HT073016 is 4.6 Mb with 50.65% G + C content and contains 4,438 genes. HT073016 also contained two plasmids named as pEM148 (Fig. 1) and pEM76 (Fig. S1). The size of pEM148 is 148,809 bp, encoding 145 genes and 2 tRNAs, with a G + C content of 44.49%. The size of pEM76 is 76,160 bp, encoding 75 genes, with a G + C content of 46.26% (Tables S1, S2). The G + C content of the plasmids is lower than that of the chromosome. Based on reads mapping, the other six E. marmotae strains also contained the two large plasmids present in HT073016.

Circular representations of the pEM148 plasmid of E. marmotae. From the outside in (to scale): circle 1 represents genes on the positive and negative strands (scale is marked in 50 kb), circle 2 shows a plot of GC content (higher values outward), and circle 3 shows a plot of GC skew (G − C)/(G + C). The red curves indicate the region to be compared. Arrows of inset indicate predicted ORFs in both strands. Shown below gene bar are locus tags. Regions in light gray indicate homologous sequences and percentages of identity between two homologous genes at the nucleotide level. The inset depicts the comparisons of the plasmid regions of T3SS, T2SS and sfp gene cluster with the corresponding regions of pCP301 of Shigella flexneri str. 301 (NC_004851), E. coli 545 chromosome (NZ_CP018976) and sfp cluster of E. coli plasmid pCFSAN004177G_03(CP012494).
Phylogenetic relationship of E. marmotae to E. coli/Shigella based on the core genome
A total of 37 bacterial genomes including 9 E. marmotae, 12 E. coli, 3 E. albertii, 1 E. fergusonii, 4 Shigella, 6 Escherichia clade I–IV, and 1 Salmonella genomes, were used to constructed a phylogenetic tree using the core genome (Table 1, Fig. 2). A total of 123,829 genes were found among these genomes, of which, 2,053 single-copy orthologs were core genes using OrthoMCL (version 2.0)16. Consistent with our previous report11, the 9 E. marmotae strains including the 2 previously designated clade V strains and 7 strains isolated from the marmots were clustered together and well separated from E. coli and Shigella. The core genome tree confirmed that E. marmotae is a different species from E. coli/Shigella.

Phylogenetic tree of E. marmotae and 30 representative genomes. The tree was constructed using the maximum likelihood algorithms in Phylip based on the core genome SNPs. Escherichia clade I–V are marked as C I to C V. C V belonged to the species E. marmotae. Values on the branch are bootstrap values out of 100 from 1000 replicates. Species and strain names are colour coded. Note that Shigella strains belong to E. coli. Salmonella Typhimurium strain LT2 is used as an outgroup.
The E. marmotae genome contains a Shigella-like type III secretion system and other Shigella virulence gene homologues
The plasmid pEM148 was found to contain a region homologous to the invasion region of the invasion plasmid pCP301 (pINV) of Shigella flexneri strain 301. The region contained 34 ORFs that share 45% to 94% identity with the type III secretion system (T3SS) of pCP301 (Fig. 1, Table S3). The genetic organization of the T3SS gene cluster of HT073016 is co-linear with that of Shigella pCP301 for nearly the entire gene cluster from virB to spa40 including the T3SS effectors for invasion and intracellular survival, assembly and function of the T3SS, and molecular chaperones (Fig. 1).
However, some genes are missing or damaged. In Shigella, ipaA, encoding an invasin, is located between acp and ipaD. In HT073016, the ipaA is truncated (1,119 bp) at the 3′ end, covering only 59% of the full length ipaA (1,902 bp), which has likely become a pseudogene. The truncated ipaA is fused with an unknown sequence to form a large mosaic gene of 4326 bp. The non-ipaA sequence shared little homology with known genes in GenBank by BLAST. Deletion of ipaA caused an ∼1000 fold decrease in the ability of S. flexneri to invade HeLa cells17. ipaJ encoding a cysteine protease that demyristoylate host proteins18 and located at the 5′ end of the Shigella invasion gene cluster is missing in HT073016. virF, encoding a master regulator of T3SS19, is also missing in E. marmotae. Another Shigella virulence gene, icsA (also known as virG), which is encoded on the Shigella pCP301 plasmid but outside the invasion region, was also missing in HT073016. IcsA mediates actin based motility inside the host cell in Shigella20.
Shigella and EIEC characteristically carry multiple copies of ipaH which encode a novel class of E3 ubiquitin ligase secreted via the T3SS21,22. We found 11 ipaH-like genes and named them as ipaH1 to ipaH11: Four on plasmid pEM148, five on plasmid pEM76, and two on the chromosome (Table S1). Seven shared homology with Shigella ipaH (Table S1), among which ipaH8 on plasmid pEM76 shared 92% identity with ipaH9.8 on Shigella pCP301 with 99% coverage. The ospC found on pEM76 shared 79% nucleotide identity with ospC4 on Shigella pCP301.
E. marmotae is capable of invading epithelial cells in vitro
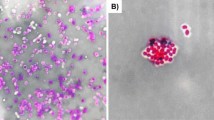
Cell invasion is a key characteristic of Shigella pathogenicity. The ability to invade epithelial cells by E. marmotae strain HT073016 was evaluated using the cell culture invasion assay, which is well established assays for Shigella23,24. In the HEp-2 cell culture invasion assay, the average number of bacteria entered into HEp-2 cells was 5 bacteria per HEp-2 cell for HT073016, in contrast, there were an average of 200 bacteria per HEp-2 cell for S. flexneri str. 301 (Fig. 3). We further quantitated the number of intracellular bacteria by colony forming unit (CFU) count. Extracellular bacteria were removed by gentamicin and intracellular bacteria were released by lysing the HEp-2 cells. The number of CFUs for E. marmotae HT073016 and S. flexneri str. 301 was 46 ± 16 and 52,000 ± 270 respectively, suggesting that there is >1000 times difference between E. Marmotae and Shigella in invasion ability. We used E. coli HB101 as negative control and no intracellular bacteria were observed by microscopy and CFU count (Fig. 3). These results indicate that E. marmotae has ability to invade epithelial cells, but its invasion ability is much lower than Shigella.

Invasion of epithelial cells by E. marmotae in vitro. HEp-2 cell invasion by E. marmotae HT073016, S. flexneri str. 301 (positive control) and E. coli HB101 (negative control) as labelled. Infected HEp-2 cells were fixed by methanol and then stained with Geimsa.
Shigella regulator virF can control E. marmotae T3SS to be responsive to temperature
The major trigger to induce the expression of the virulence genes of the Shigella invasion plasmid is a temperature shift to 37 °C before Shigella enters the host25. virF, as a global positive regulator, is responsible for triggering T3SS expression in Shigella26. However, no virF homologue was found in E. marmotae HT073016. We examined the response of T3SS of E. marmotae HT073016 to temperature shift by measuring T3SS ipaD expression. The recA gene was used as a reference. The ipaD expression of E. marmotae HT073016 at 25 °C and 37 °C was almost the same and was considerably lower than S. flexneri str. 301 at 25 °C (Fig. 4). In contrast, the ipaD expression of S. flexneri str. 301 was responsive to temperature shift with 382 times higher expression at 37 °C than 25 °C. Therefore, unlike Shigella, the expression of T3SS of E. marmotae HT073016 is not responsive to temperature modulation. We then cloned the virF gene from S. flexneri str. 301 with its promoter region into HT073016 to determine whether Shigella virF can regulate E. marmotae T3SS expression. The ipaD gene expression of recombinant HT073016 (virF+) was increased by 73 folds, when the temperature shifted from 25 °C to 37 °C (Fig. 4), demonstrating that Shigella virF can exert control on the expression of E. marmotae T3SS. We further tested the recombinant HT073016 (virF+) for its invasion ability (Fig. S2). The number of intracellular bacteria (CFUs) for the virF transformant was 198 ± 66 in comparison to the CFU of 46 ± 16 for the wild type. The difference is statistically significant (t test, p < 0.01).

Expression of ipaD under different temperature conditions as determined by qRT-PCR. Total RNA was harvested from HT073016, HT073016(virF+) and S. flexneri strain 301 cultivated at 25 °C and 37 °C. Relative transcript levels were calculated using the ∆∆CT method and fold changes in comparison to HT073016 at 37 °C. All values have been normalized to the endogenous reference gene recA. Means and standard deviations stand for three independent experiments were shown with * being p < 0.05 in the indicated comparisons. Error bars are +SD.
The E. marmotae genome contained virulence genes of other enteric bacteria
There are four singularly located virulence genes found in pEM148: An enterohaemorrhagic E. coli (EHEC) epeA, E. coli lifA/efa (lymphocyte inhibitory factor A), cif (cycle inhibiting factor) and Yersinia yopJ homologue. epeA encodes a serine protease belonging to the family of serine protease autotransporters of Enterobacteriaceae (SPATEs)27. Shigella sepA also belongs to SPATES family27 and is absent in E. marmotae. E. marmotae lifA/efa shares 80% identity with E. coli lifA/efa which has a dual role in suppressing cytokine expression and functioning as an adhesin28,29. The E. marmotae cif shares 74% identity with E. coli cif which encodes a T3SS effector that induces host cell cycle arrest and reorganization of the actin cytoskeleton30. The E. marmotae yopJ shares 82% identity with Yersinia yopJ, which encodes an acetyl transferase targeting and inhibiting host immune cells31 (Table S4).
There are four gene clusters associated with virulence. A type II secretion system (T2SS) is present on plasmid pEM148 which shares an overall 91% identity with those of E. coli and Shigella (Fig. 1). However, the E. coli and Shigella T2SS was located on the chromosome32. A sorbitol-fermenting fimbriae protein (Sfp) locus is present on pEM148 which contained seven genes and shared 88% identity with that of E. coli (Fig. 1). The sfp locus is associated with adhesion to host cells in EHEC33. In addition to plasmid encoded virulence genes, HT073016 contains 2 type VI secretion systems (T6SS) on the chromosome (Table S5), one of which, T6SS1, is more homologous to that of Salmonella, while the other (T6SS2) is more similar to the one in E. coli O42 (Fig. S3). However, T6SS2 appears to be partial as the majority of the E. coli T6SS homologs were absent (Table S5).
The E. marmatae genome contained numerous copies of IS1414 that encodes an enterotoxin
The E. marmotae genome also contained a large number of ISs (121) (Table 1). The predominant species are IS1414 (37), IS4 (36), followed by ISCro1 (24) and ISEc16 (19). IS1414 is found on both the chromosome (32 copies) and the two plasmids (5 copies) (Tables 2, S6). Interestingly IS1414 carries a heat-stable enterotoxin gene astA inside the IS as previously found in E. coli34,35. IS1414 is found at a much smaller number from 1 to 8 copies in E. coli. No IS1414 was found in the S. flexneri str. 301 genome.
E. marmotae carries the same O antigen gene cluster as Shigella dysenteriae type 8 and E coli O38
All seven E. marmotae strains carry the same O antigen. The E. marmotae O-antigen biosynthesis gene cluster shares 99.41% identity with that of E. coli O38 and 99.24% with that of Shigella dysenteria type 8 (SD8) which is much higher than chromosomal genes between the HT073016 and Shigella at around 60%, suggesting that one or all acquired the O antigen gene cluster from the same source recently (Fig. S4). Slide agglutination test confirmed that E. marmotae HT073016 agglutinated with SD8 diagnostic antiserum for Shigella serotyping.
Discussion
Shigella evolved from commensal E. coli through acquisition of key virulence genes primarily carried by the invasion plasmid pINV which is shared by Shigella and EIEC13. There had been no reports of any other species carrying a similar plasmid. In this study we show that E. marmotae strains isolated from wild rodents from the Qinghai-Tibet plateau contained a plasmid that carried a T3SS gene cluster other virulence genes with high homology to that on the pINV, and also virulence genes on the chromosome. E. marmotae was also found to be invasive to HEp-2 cells. Thus, E. marmotae is a potential invasive pathogen. E. marmotae also shared the multicopy ipaH gene with Shigella and EIEC, which participates to modulate the immune response of the host36.
Shigella and EIEC arose within E. coli multiple times independently by gaining a similar invasion plasmid37. However, no Shigella-like plasmid or Shigella-like T3SS system in other species had been reported previously. The finding that E. marmotae gained a similar T3SS and other invasion related genes showed parallel evolution of the Shigella-like T3SS and associated virulence genes within the genus Escherichia. Firstly, the T3SS gene cluster showed co-linearity over the entire gene cluster between E. marmotae and Shigella and both T3SS were carried by a plasmid, although the two plasmids shared little similarity in other regions. Secondly, most of the key Shigella T3SS effectors secreted are present in E. marmotae.
Cell invasion experiments showed that E. marmotae was much less invasive than Shigella. The absence of three important pathogenic mechanisms: temperature modulation and IcsA-based intracellular motility and absence of ipaA may have contributed to this difference. Therefore E. marmotae is a less invasive pathogen. The master regulator VirF is absent in E. marmotae. VirF responds to a diverse range of environmental signals26. Our experiments showed that introduction of Shigella virF with its promoter region into E. marmotae rendered the E. marmotae T3SS gene expression responsive to temperature because the virF gene from Shigella contain its promoter region which responds to temperature and also increased the invasiveness to HEp-2 cells. E. marmotae lacked IcsA for actin based motility which is used for movement within and between epithelial cells. For Shigella, the key virulence factors were all encoded on pINV. It has been shown that pINV is a composite plasmid originated from different sources14. For E. marmotae, the plasmid pEM148 is also a composite plasmid with G + C content of the invasion region (T3SS gene cluster) differed from the other regions of the plasmid. E. marmotae is yet to acquire other key virulence genes such as icsA to develop full blown Shigella-like pathogenicity.
In addition to the virulence genes similar to those of Shigella, the E. marmotae genome also contained other virulence genes. These include E. coli homologues, lifA/efa, astA, cif, and the sfp gene cluster, Yersinia yopJ, one T2SS, and two T6SSs. It is particularly interesting that the astA gene which is embedded entirely within IS1414 is present in E. marmotae with 37 copies. astA encodes heat-stable enterotoxin 1 (EAST1) which was initially discovered in enteroaggregative E. coli34. Based on the predominance of IS1414 in E. marmotae and its low frequency in E. coli, it is highly likely that the E. coli IS1414 and its associated astA gene was originated from E. marmotae. Escherichia Clade V strain TW09308, genetically a member of E. marmotae11, also contained IS1414 and the astA gene38. Escherichia Clade V strains have been found in the environment including fresh water and marine sediments and from different geographical regions including Australia, the US and Italy38. The environmental Escherichia Clade V strains from the marine sediments were shown to carry the astA gene and were also found to be adhesive to human epithelial cells39. Therefore E. marmotae may contain both widely distributed environmental strains (ie. Escherichia Clade V36) and animal intestinal strains such as those isolated from M. himalayana with differences in pathogenicity. E. marmotae is likely to be an environmental organism turning into a pathogen.
However, the 7 E. marmotae strains we isolated were from apparently healthy marmots. It remains to be demonstrated whether E. marmotae can cause disease in humans or other animals. The genome content of virulence genes and our cell invasion experiments suggest that E. marmotae is likely to be an invasive pathogen. Further studies will be required to demonstrate its pathogenicity and disease-causing potential to humans.
This study further supports that wild marmots are a reservoir of potential human pathogens and virulence genes. We previously sampled E. coli from M. himalayana and the majority of the 112 E. coli strains sequenced carried multiple virulence genes and in particular they carried virulence genes from different pathogenic types of E. coli as potential hybrid pathogens10. Qinghai-Tibet plateau is a relatively pristine environment. The area is sparsely populated and human activity is low, due to its high altitude (2,700 to 5,450 meters above sea level) and harsh climate in winter which make it less suitable for human inhabitation. There are many wild animal species in the Qinghai-Tibet plateau. Therefore, it is likely that other wild animals also carry E. marmotae and other known or unknown pathogens.
In conclusion, we characterised E. marmotae strains isolated from wild marmots and showed that E. marmotae acquired a plasmid that carries a Shigella-like T3SS system. It also carried many homologues of Shigella effectors. However, cell invasion assays showed that E. marmotae is far less invasive than Shigella, which may be due to that E. marmotae lacks the VirF mediated regulatory system to be responsive to environmental changes and the Shigella IcsA-actin based intracellular motility. This study also provides further support that wild animals are reservoirs of potential novel human pathogens.
Materials and Methods
Bacteria strains, cell culture and media
E. coli HB101, S. flexneri str. 301, E. marmotae strains HT073016, HT080709, HT080711, HT072503, HT073105, HT080118 and HT080401 were grown in Luria-Bertani (LB) broth or on LB agar at 37 °C. HEp-2 cells were cultured in DMEM (Gibco) with 10% bovine fetal calf serum.
Genome sequencing, annotation and whole genome phylogenetic analysis
DNA was prepared with the Wizard Genomic DNA Purification Kit (Promega, USA). Seven isolates were sequenced using Illumina HiSeq2000 by Beijing Genomics Institute. HT073016 was further sequenced using Pacific Biosciences RSII DNA sequencing system (Pacific Biosciences, Menlo Park, CA, USA) by Tianjin Biochip Corporation. De novo assembly of the insert reads of Pacific Biosciences SMRT sequencing was performed with the Hierarchical Genome Assembly Process (HGAP_Assembly.2) algorithm in SMRT Portal (version 2.3.0). Circularization was achieved by manual comparison and removal of a region of overlap, and the final genome was confirmed by remapping of sequence data. Initial annotation of the genome was done using the Rapid Annotation using Subsystem Technology online interface, and further annotated by BLASTP and BLASTN against NCBI’s conserved domain database and non-redundant databases. Virulence gene annotations were recovered using VFDB (http://www.mgc.ac.cn/VFs/download.htm). Plasmids carrying virulence genes were then selected for a more in-depth annotation. Each CDS initially annotated as transposase was further annotated by BLASTN against the ISFINDER database. Insertion sequence terminal inverted repeats (TIR) and direct repeats (DR) were identified using comparisons with known published elements. Schematic map of plasmids and gene organization diagrams were drawn with in-house Perl scripts and Inkscape40. The phylogenetic tree based on the core genome SNPs was constructed using the maximum likelihood algorithms in PHYLIP (http://evolution.gs.washington.edu/phylip.html). Bootstraps were performed with 1,000 replicates. The resulting phylogeny was displayed by SplitsTree (http://en.bio-soft.net/tree/SplitsTree.html), and edited by iTOL and Adobe illustrator.
GenBank accession number of genomes sequenced in this work is SRS2488458 - SRS2488464 (Bioproject identification number PRJNA401298), CP025979- CP025981.
Epithelial cell invasion assay
HEp-2 cells were seeded at 1 × 105 cells/well into 24-well cell culture plates (Costar, Corning) and cultured overnight. Cells were washed three times with pre-warmed DMEM to remove the antibiotics and serum before addition of bacteria. Subconfluent monolayers of HEp-2 cells were infected with approximately 2 × 106 exponential-phase bacteria in DMEM at 37 °C. Following an initial invasion period of 1 h, cells were washed three times by DMEM, and the infection was allowed to continue for an additional 1 h after the addition of 100 μg/ml gentamicin, which kills extracellular but not intracellular bacteria. The infected HEp-2 cells were washed, followed by fixation with methanol and then stained with Giemsa for analysis under a light microscope (Nikon ECLIPSE 80i). The invasive capacity of bacteria was further measured by counting the viable number of internalized bacteria using colony forming unit (CFU) count method on LB agar plate after the cells were lysed by 0.25% Triton X-100 to release the bacteria. The S. flexneri str. 301, and E. coli HB101 were used as positive and negative control, respectively.
Recombinant strain construction
First, we amplified the DNA region containing S. flexneri str. 301 virF gene and its promoter using the primers (virF-promoter-F: 5′-AGAAGCTGCATAAGCTCTTTCTTC-3′; virF-promoter-R: 5′-GGGGAAAACCCATCTGGCAA-3′). The PCR product was purified by Gel Extraction Kit (Qiagen). Thereafter the purified product was cloned into pMD18-T vector (Takara) and transformed into E. coli JM109 for amplification. Eventually after extracted by Plasmid Mini Kit (Omega), the recombinant plasmid was transformed into competent E. marmotae HT073016 cell to construct E. marmotae HT073016(virF+).
Quantitative Real-time PCR assay
E. marmotae HT073016, HT073016(virF+) and S. flexneri str. 301 were cultured in the LB medium at 37 °C and 25 °C with constant shaking until reached the exponential phase of growth. Then the total RNA of each sample was extracted by using the RNeasy Mini kit (Qiagen) and digested by TURBO DNase (Ambion) and reversely transcribed to cDNA by PrimeScript™ RT Master Mix (Takara). RNA concentrations were determined by a NanoDrop spectrophotometer. Transcriptional levels of the ipaD gene from E. marmotae HT073016, HT073016(virF+) and S. flexneristr. 301 were measured by the quantitative real-time PCR. Primers for ipaD and reference gene recA were listed as followed (ipaD-F: 5′-GATAATGCAAAATATCAGGCATGGAA-3′, ipaD-R: 5′-CATGAGCTTATTGTACTACTCAAAACCTT-3′; recA-F: 5′-ACAAACAGAAAGCGTTGGCG-3′, recA-R: 5′-CCAAGCGCGATATCCAGTGA-3′). Reactions were prepared using SYBR Premix Ex TaqTM II (Takara) in a total volume of 25 μL. The real-time PCR assays were performed by using Rotor-Gene Q (Qiagen) following the amplification program: 45 cycles at 95 °C for 5 sec, 60 °C for 30 sec and 72 °C for 30 sec. For each sample, the raw real-time PCR data for the target gene ipaD were normalized against the reference gene recA, and fold changes were calculated using the △△CT method as reported by Livak and Schmittgen41. The results were based on three individual experiments.
References
Daszak, P., Cunningham, A. A. & Hyatt, A. D. Emerging Infectious Diseases of Wildlife–Threats to Biodiversity and Human Health. Science 287, 443 (2000).
Neumann, G., Chen, H., Gao, G., Shu, Y. & Kawaoka, Y. H5N1 influenza viruses: outbreaks and biological properties. Cell Res 20, 51–61, https://doi.org/10.1038/cr.2009.124 (2010).
Burgdorfer, W. Discovery of the Lyme disease spirochete and its relation to tick vectors. The Yale journal of biology and medicine 57, 515–520 (1984).
Steppan, S. et al. Molecular phylogeny of the marmots (Rodentia: Sciuridae): tests of evolutionary and biogeographic hypotheses. Syst Biol 48, 715–734, https://doi.org/10.1080/106351599259988 (1999).
Qian, Q. et al. Mapping risk of plague in Qinghai-Tibetan Plateau, China. BMC Infect Dis. 14, https://doi.org/10.1186/1471-2334-14-382 (2014).
Hu, S. et al. Helicobacter himalayensis sp. nov. isolated from gastric mucosa of Marmota himalayana. International journal of systematic and evolutionary microbiology 65, 1719–1725, https://doi.org/10.1099/ijs.0.000163 (2015).
Niu, L. et al. Streptococcus halotolerans sp. nov. isolated from the respiratory tract of Marmota himalayana in Qinghai-Tibet Plateau of China. International journal of systematic and evolutionary microbiology 66, 4211–4217, https://doi.org/10.1099/ijsem.0.001337 (2016).
Niu, L. et al. Streptococcusmarmotae sp. nov., isolated from the respiratory tract of Marmota himalayana. International journal of systematic and evolutionary microbiology 66, 4315–4322, https://doi.org/10.1099/ijsem.0.001350 (2016).
Niu, L. et al. Streptococcus himalayensis sp. nov., isolated from the respiratory tract of Marmota himalayana. International journal of systematic and evolutionary microbiology 67, 256–261, https://doi.org/10.1099/ijsem.0.001609 (2017).
Lu, S. et al. Insights into the evolution of pathogenicity of Escherichia coli from genomic analysis of intestinal E. coli of Marmota himalayana in Qinghai-Tibet plateau of China. Emerging microbes & infections 5, e122, https://doi.org/10.1038/emi.2016.122 (2016).
Liu, S. et al. Escherichia marmotae sp. nov., isolated from faeces of Marmota himalayana. International journal of systematic and evolutionary microbiology 65, 2130–2134, https://doi.org/10.1099/ijs.0.000228 (2015).
Philpott, D. J., Edgeworth, J. D. & Sansonetti, P. J. The pathogenesis of Shigella flexneri infection: lessons from in vitro and in vivo studies. Philos Trans R Soc Lond B Biol Sci 355, 575–586 (2000).
Lan, R. & Reeves, P. R. Eschericheria coli in disguise: molecular origins of Shigella. Microbes and Infection 4, 1125–1132 (2002).
Buchrieser, C. et al. The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Molecular Microbiology 38, 760–771 (2000).
Venkatesan, M. M. et al. Complete DNA sequence and analysis of the large virulence plasmid of Shigella flexneri. Infection and Immunity 69, 3271–3285 (2001).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome research 13, 2178–2189, https://doi.org/10.1101/gr.1224503 (2003).
VanNhieu, G. T., BenZeev, A. & Sansonetti, P. J. Modulation of bacterial entry into epithelial cells by association between vinculin and the Shigella IpaA invasin. Embo Journal 16, 2717–2729, https://doi.org/10.1093/emboj/16.10.2717 (1997).
Burnaevskiy, N. et al. Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor Ipa. J. Nature 496, 106–109, http://www.nature.com/nature/journal/v496/n7443/abs/nature12004.html#supplementary-information (2013).
Tobe, T. et al. Temperature-regulated expression of invasion genes in Shigella flexneri is controlled through the transcriptional activation of the virB gene on the large plasmid. Mol Microbiol 5, 887–893 (1991).
Bernardini, M. L., Mounier, J., d’Hauteville, H., Coquis-Rondon, M. & Sansonetti, P. J. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci USA 86, 3867–3871 (1989).
Rohde, J. R., Breitkreutz, A., Chenal, A., Sansonetti, P. J. & Parsot, C. Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell host & microbe 1, 77–83, https://doi.org/10.1016/j.chom.2007.02.002 (2007).
Tran Van Nhieu, G. et al. Actin-based confinement of calcium responses during Shigella invasion. Nature communications 4, 1567, https://doi.org/10.1038/ncomms2561 (2013).
Lin, F. C., Yang, T. L. & Wu, P. F. Experimental keratoconjunctivitis shigellosis in Guinea Pigs. II. Histologic findings. Chinese medical journal 83, 463–469 (1964).
DuPont, H. L., Levine, M. M., Hornick, R. B. & Formal, S. B. Inoculum size in shigellosis and implications for expected mode of transmission. The Journal of infectious diseases 159, 1126–1128 (1989).
Konkel, M. E. & Tilly, K. Temperature-regulated expression of bacterial virulence genes. Microbes Infect 2, 157–166 (2000).
Dorman, C. J. & Porter, M. E. The Shigella virulence gene regulatory cascade: a paradigm of bacterial gene control mechanisms. Molecular Microbiology 29, 677–684, https://doi.org/10.1046/j.1365-2958.1998.00902.x (1998).
Dautin, N. Serine protease autotransporters of enterobacteriaceae (SPATEs): biogenesis and function. Toxins 2, 1179–1206, https://doi.org/10.3390/toxins2061179 (2010).
Klapproth, J. M. et al. Citrobacter rodentium lifA/efa1 is essential for colonic colonization and crypt cell hyperplasia in vivo. Infect Immun 73, 1441–1451, https://doi.org/10.1128/iai.73.3.1441-1451.2005 (2005).
Badea, L. et al. Contribution of Efa1/LifA to the adherence of enteropathogenic Escherichia coli to epithelial cells. Microbial pathogenesis 34, 205–215 (2003).
Marches, O. et al. Enteropathogenic and enterohaemorrhagic Escherichia coli deliver a novel effector called Cif, which blocks cell cycle G2/M transition. Mol Microbiol 50, 1553–1567 (2003).
Mukherjee, S. et al. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312, 1211–1214, https://doi.org/10.1126/science.1126867 (2006).
Yang, F. et al. Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res 33, 6445–6458, https://doi.org/10.1093/nar/gki954 (2005).
Brunder, W., Khan, A. S., Hacker, J. & Karch, H. Novel type of fimbriae encoded by the large plasmid of sorbitol-fermenting enterohemorrhagic Escherichia coli O157:H(−). Infect Immun 69, 4447–4457, https://doi.org/10.1128/iai.69.7.4447-4457.2001 (2001).
Menard, L. P. & Dubreuil, J. D. Enteroaggregative Escherichia coli heat-stable enterotoxin 1 (EAST1): a new toxin with an old twist. Critical reviews in microbiology 28, 43–60, https://doi.org/10.1080/1040-840291046687 (2002).
McVeigh, A. et al. IS1414, an Escherichia coli insertion sequence with a heat-stable enterotoxin gene embedded in a transposase-like gene. Infect Immun 68, 5710–5715 (2000).
Ashida, H. & Sasakawa, C. Shigella IpaH Family Effectors as a Versatile Model for Studying Pathogenic Bacteria. Frontiers in cellular and infection microbiology 5, 100, https://doi.org/10.3389/fcimb.2015.00100 (2015).
Lan, R., Alles, M. C., Donohoe, K., Martinez, M. B. & Reeves, P. R. Molecular evolutionary relationships of enteroinvasive Escherichia coli and Shigella spp. Infect Immun 72, 5080–5088, https://doi.org/10.1128/IAI.72.9.5080-5088.2004 (2004).
Luo, C. et al. Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc Natl Acad Sci USA 108, 7200–7205, https://doi.org/10.1073/pnas.1015622108 (2011).
Vignaroli, C. et al. Adhesion of marine cryptic Escherichia isolates to human intestinal epithelial cells. The ISME journal 9, 508–515, https://doi.org/10.1038/ismej.2014.164 (2015).
Yuan, S., Chan, H. C., Filipek, S. & Vogel, H. PyMOL and Inkscape Bridge the Data and the Data Visualization. Structure 24, 2041–2042, https://doi.org/10.1016/j.str.2016.11.012 (2016).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols 3, 1101–1108 (2008).
Acknowledgements
This work was supported by the national key research and development plan, 2016YFC1201903), National Natural Science Foundation of China (81290345, 81471918), the Ministry of Science and Technology of China (Mega Project of Research on The Prevention and Control of HIV/AIDS, Viral Hepatitis Infectious Diseases, 2013ZX10004-101),and Chinese Academy of Medical Sciences (2018RC310026).
Author information
Authors and Affiliations
Contributions
J.X., S.L. and R.L. designed the study; S.L., J.P., X.X., S.L., J.Y., D.J., X.M., X.L., H.S. Y.X. and C.Y. performed the experiments; S.L., J.F., Y.W., X.D. and R.L. analyzed the data; S.L., J.F., R.L. and J.X. wrote the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41598_2019_46831_MOESM1_ESM.pdf
Genomic and molecular characterisation of Escherichia marmotae from wild rodents in Qinghai-Tibet plateau as a potential pathogen
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, S., Feng, J., Pu, J. et al. Genomic and molecular characterisation of Escherichia marmotae from wild rodents in Qinghai-Tibet plateau as a potential pathogen. Sci Rep 9, 10619 (2019). https://doi.org/10.1038/s41598-019-46831-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-46831-3
This article is cited by
-
Metagenomic analysis of viromes in tissues of wild Qinghai vole from the eastern Tibetan Plateau
Scientific Reports (2022)
-
Chromosome-encoded IpaH ubiquitin ligases indicate non-human enteroinvasive Escherichia
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
