
Abstract
Wastewater treatment is crucial to environmental hygiene in urban environments. However, wastewater treatment plants (WWTPs) collect chemicals, organic matter, and microorganisms including pathogens and multi-resistant bacteria from various sources which may be potentially released into the environment via WWTP effluent. To better understand microbial dynamics in WWTPs, we characterized and compared the bacterial community of the inflow and effluent of a WWTP in Berlin, Germany using full-length 16S rRNA gene sequences, which allowed for species level determination in many cases and generally resolved bacterial taxa. Significantly distinct bacterial communities were identified in the wastewater inflow and effluent samples. Dominant operational taxonomic units (OTUs) varied both temporally and spatially. Disease associated bacterial groups were efficiently reduced in their relative abundance from the effluent by the WWTP treatment process, except for Legionella and Leptospira species which demonstrated an increase in relative proportion from inflow to effluent. This indicates that WWTPs, while effective against enteric bacteria, may enrich and release other potentially pathogenic bacteria into the environment. The taxonomic resolution of full-length 16S rRNA genes allows for improved characterization of potential pathogenic taxa and other harmful bacteria which is required to reliably assess health risk.
Similar content being viewed by others

Introduction
Drinking water is a critical resource for which it is challenging to maintain hygiene in urban areas under persistent anthropogenic influence1,2,3,4. Pollutants and antibiotic resistant bacteria are constantly released by wastewater treatment plants (WWTP) into the environment which can result in human and (aquatic) animal health risk5,6,7,8,9,10,11. Treated sewage is also a major source of human-derived bacteria in the urban water environment, including potential pathogens that may survive the treatment process. Sewage inflow partly reflects the bacterial community of humans12,13. In two WWTPs in Hong Kong (China) pathogenic bacteria such as Clostridium perfringens, Legionella pneumophila and Mycobacterium tuberculosis like species were found to be common14. Thus, the WWTP effluent may not be completely depleted of (human) pathogens and the microbiome of the effluent and its nutrients may even promote the growth and proliferation of pathogenic bacteria in the environment. For example, Wakelin et al.15 demonstrated that constant effluent input in combination with increased nutrient levels in the sediment downstream of a WWTP in Australia affected the bacterial community in the sediment substantially and increased the overall diversity. In addition, a study in rural Bangladesh revealed anthropogenic contamination of groundwater pumped from shallow tubewells with faecal bacteria from the genera Shigella and Vibrio16 indicating the potential risk of faecal contamination of the natural environment via anthropogenic effluents.
WWTPs are considered hotspots for antibiotic resistant genes and for the spread of bacteria into the environment9,17. The presence of antibiotic resistant bacteria also increases the potential risk of gene transfer to non-resistant bacteria18,19,20. Several environmental bacteria are prone to developing multidrug resistance such as Acinetobacter spp., Aeromonas spp. and Pseudomonas spp.21,22,23. Adapted to humid and various aquatic environments24,25,26,27,28,29,30,31 these human-derived bacteria are part of the microbial communities in municipal WWTPs32,33,34. For example, an increase of antibiotic resistant Acinetobacter spp. in WWTPs has been shown by Zhang et al.21.
In addition, bacterial communities of wastewater include members of different taxonomic, biochemical (e.g. N2-fixation, nitrification, denitrification, sulphur oxidation) and physiological (e.g. anaerobic, aerobic, phototrophic, heterotrophic) groups, of which many provide functional advantages for water cleaning, such as nutrient removal. However, these communities also contain bacteria of human and animal origin which may interact with the bacterial communities of natural waters (e.g. rivers and lakes) in unpredictable ways. There are few studies comparing WWTP bacterial communities in inflow and effluent with the few undertaken restricted to a few countries, i.e. the USA, Hong Kong (China), and Spain5,12,13,35,36. Furthermore, these studies have provided relatively little taxonomic resolution since molecular identification has been limited to short hypervariable regions of the 16S rRNA gene due to amplicon size constraints in sequencing on the Illumina or Roche 454 platforms5,12,13,35,36. Providing only a restricted phylogenetic resolution, these methods do not allow for the reliable identification of human pathogenic bacteria in the environment. Full 16S rRNA gene sequencing, however, can provide improved taxonomic identification on the genus and species level. Therefore, we used single molecule real time (SMRT) sequencing (PacBio® Sequel platform) to determine full-length bacterial 16S rRNA gene PCR amplicon sequences in order to improve the characterization of bacterial communities in wastewater samples and to compare the communities between inflow and effluent samples. Inflow and effluent samples from a WWTP in Berlin (Germany) were collected every three months for one year to characterize in detail and compare the bacterial communities including potentially pathogenic bacteria.
Results
Bacterial community composition and dominant OTUs
Using an OTU clustering cut-off of 99% sequence similarity, we were able to identify a total of 7,068 OTUs (initial data) of which 3,860 were left after rarefaction. Beta-diversity analyses were performed on a rarefied OTU-count table. The rationale was to describe the main changes in microbial community composition and the sequence coverage of our samples (ranging from 4,468 to 78,350 reads) did not allowed for a robust analysis of the “rare-biosphere” for which 0.1–0.2 M reads per sample are recommended37. As expected, the numbers of reads and OTUs were reduced after rarefaction (Supplementary Fig. S1). Nevertheless, the rarefied dataset was highly representative of the original non rarefied data (R = 0.99, p-value < 0.001) as confirmed by a Mantel correlation test based on the Bray-Curtis dissimilarity and Person coefficient.
Fingerprinting techniques have shown that the 10–50 most abundant taxa usually contribute more than 0.1–1.0% to the total cell counts. That is why, in concordance with other studies38,39,40, bacterial phyla and genera representing more than 1.0% of the total community are considered dominant taxa.
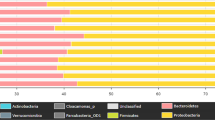
Predominant phyla in the inflow were Firmicutes, Proteobacteria, Bacteroidetes and Actinobacteria with an average abundance of 52.2 ± 4.4%, 37.8 ± 4.7%, 4.9 ± 1.9%, and 2.2 ± 0.2%, respectively. In contrast, the effluent was dominated by Proteobacteria (54.8 ± 3.3%), Bacteroidetes (15.7 ± 1.1%), Firmicutes (14.3 ± 5.0%), Planctomycetes (2.9 ± 1.1%), Actinobacteria (2.6 ± 0.4%), Verrucomicrobia (2.1 ± 0.4%) and Acidobacteria (1.3 ± 0.4%) (Fig. 1). Families of the dominant phyla Proteobacteria, Bacteroidetes and Firmicutes contributing more than 1% in at least one sample are shown in Fig. 2. Firmicutes were dominated by Acidaminococcaceae, Enterococcaceae, Eubacteriaceae, Lachnospiraceae, Peptostreptococcaceae, Ruminococcaceae, Streptococcaceae, Veillonellaceae, Christensenellacea and Clostridiaceae 1 with Lachnospiraceae and Ruminococcaceae as most abundant. Proteobacteria were mainly represented by the families Aeromonadaceae, Comamonadaceae, Enterobacteriaceae, Moraxellaceae, Neisseriaceae, Rhodobacteraceae, Rhodocyclaceae Campylobacteriaceae and Xanthomonadaceae. For the phylum Bacteroidetes, the families Bacteroidaceae, Chitinophagaceae, Cytophagaceae, Flavobacteriaceae, Porphyromonadaceae, Prevotellaceae, Rikenellaceae and Saprospiraceae were dominant.

Relative abundance of dominant bacterial phyla (contributing more than 1% to the total bacterial community) in inflow and effluent samples after data rarefaction. (A) Bar charts show the percentage bacterial taxonomic composition for each sample. (B) Boxplots showing the average relative abundance of the dominant bacterial phyla between inflow and effluent. The p-value (p) indicates the significance of the differences based on a PERMANOVA with p < 0.05 being significant.

Average relative abundance of families contributing more than 1.0% to the total bacterial community in inflow and/or effluent. Only families of the three most abundant phyla Proteobacteria, Bacteroidetes and Firmicutes are shown.
OTUs contributing more than 1% to the total bacterial community in at least one sample are listed in Table 1. In addition, OTUs that significantly differ between inflow and effluent are marked in Table 1 and belong to multiple genera: Canditadus Accumulibacter, Candidatus Competibacter, Comamonadaceae unclassified, Dechloromonas, Nitrosomonas, Nitrospira, Paracoccus, Rhodoferax, unclassified Run-SP154, Simplicispira, Streptococcus, unclassified Saprospiraceae and Uruburuella. Using full-length 16S rRNA reads, we were able to reliably identify many OTUs at high taxonomic resolution (often at species level) by comparing them with reference sequences from known bacterial species (Table 1, Table 2) based on a global SILVA alignment (SINA Aligner) for rRNA genes41.
A comparison of the PacBio generated OTU sequences with short-read sequences, which were generated by extracting a 477 bp fragment of the hypervariable regions V3-V4 according to the primer pair of Klindworth et al.42 from the PacBio reads, was performed for the genus Acinetobacter as an example. Out of 113 OTUs related to the genus Acinetobacter 18 could be resolved to the species level when using full-length PacBio reads and 10 when using the hypervariable region V3-V4. The bootstrap values were always lower when using shorter sequences. Two OTUs yielded different phylogenetic results depending on sequence length (Supplementary Table S1, Fig. S2).
Inflow versus effluent samples and dominant OTUs
Principal coordinate analysis defined two main clusters: inflow and effluent samples (Fig. 3). Within the inflow cluster the samples from April and October were most similar to each other, whereas in the effluent cluster February and April or July and October samples were clustered more closely together. A permutational multivariate analysis of variance (PERMANOVA) revealed a significant difference between inflow and effluent samples with a p-value of 0.02. Furthermore, the dominant bacterial phyla Bacteroidetes, Firmicutes, Planctomycetes and Verrucomicrobia differed significantly in their relative abundance between inflow and effluent samples, whereas the phylum Actinobacteria did not show a significant difference between both sample groups (Fig. 1).

Principal coordinate analysis (PCoA) of the bacterial community based on Bray-Curtis similarity. Inflow and effluent samples are defined as circles and squares, respectively. Different sampling time points are indicated by blue colour for February, green colour for April, red colour for July and brown colour for October.
Phylogenetic analysis of genera that contain known pathogens
The advantage of full-length 16S rRNA gene sequencing was that, with some restrictions, more refined and reliable taxonomic assignment, even to the species level, was possible. While most of the previous studies used only the information of certain hypervariable regions of the 16S rRNA, we were able to use all phylogenetically relevant sites of the whole 16S rRNA gene. We attempted to identify OTUs to a higher taxonomic level (e.g. species level), focusing on bacterial groups known to contain strains relevant for human health (Table 2). The analysis was carried out using maximum likelihood based phylogenetic approaches and including reference sequences from the SILVA database43,44. Three major groups of OTUs were identified representing (1) waterborne/-transmitted bacteria (i.e., Legionella, Leptospira, Vibrio and Mycobacterium)45,46,47,48, (2) enteric bacteria (i.e., Campylobacter, Clostridium, Salmonella, Shigella and Yersinia)49,50,51,52,53,54, and (3) environmental bacteria (i.e. Acinetobacter, Aeromonas and Pseudomonas) that include important nosocomial pathogens, which can also acquire multi-drug resistance55,56,57,58,59.
Waterborne bacteria: Legionella, Leptospira, Mycobacterium and Vibrio
Legionella spp. and Leptospira spp. contributed to up to 0.9% and 1.0% to the bacterial community after rarefaction, respectively with increasing numbers from inflow to effluent (Fig. 4). Identified OTUs were closely related to Legionella lytica, L. feeleii (Supplementary Fig. S3) and Leptospira alstonii (Supplementary Fig. S4). The genus Mycobacterium was only present in the October effluent samples with a relative abundance of 0.02%, whereas Vibrio was not detected in either the inflow or the effluent (Fig. 4).

Relative abundance (after rarefaction) of genera with known potential pathogens. They were grouped in environmental, waterborne and enteric, and are shown for each sample of inflow and effluent.
Enteric bacteria: Campylobacter, Clostridium, Escherichia/Shigella, Salmonella and Yersinia
Campylobacter and Salmonella spp. were not detectable. The genus Clostridium (sensu-stricto) contributed between 0.1–0.9% to the bacterial community. Escherichia/Shigella and Yersinia decreased from inflow to effluent in relative abundance with Yersinia spp. being absent from the effluent samples (Fig. 4). According to our phylogenetic analyses probable species are Clostridium perfringens, C. botulinum, C. butyricum (Supplementary Fig. S5), Yersinia massiliensis, Y. frederiksenii, Y. enterocolitica and Y. media (Supplementary Fig. S6). OTUs from the Escherichia/Shigella group did not show clear sequence similarity with any known species.
Environmental bacteria: Acinetobacter, Aeromonas and Pseudomonas
The genera Acinetobacter, Aeromonas and Pseudomonas were present in all samples, but their relative abundance decreased from inflow water to effluent in each of the sampled months (Fig. 4). Acinetobacter and Aeromonas spp. represented up to 9.5% and 5.8% of the bacterial community in the inflow, but only up to 1.3% and 1.1% in the effluent, respectively, while Pseudomonas spp. contributed only between 0.02% and 0.5% to the total bacterial community decreasing from inflow to effluent. OTUs were closely related to the described species Acinetobacter beijerinckii, A. haemolyticus, A. baumannii (Supplementary Fig. S7), Aeromonas sharmana, A. media (Supplementary Fig. S8), Pseudomonas alcaligenes and P. aeruginosa (Supplementary Fig. S9).
Discussion
The advantage of full-length 16S rRNA gene sequencing was that, with some restrictions, more refined and reliable taxonomic assignment, even to the species level, was possible. While most of the previous studies used only the information of certain hypervariable regions of the 16S rRNA, we were able to use all phylogenetically relevant sites of the whole 16S rRNA gene. Huse et al.60 compared full-length sequence with V3 and V6 hypervariable regions and found both methods could resolve the taxonomy similarly at the level of genus. Our genus comparison of Acinetobacter demonstrated better species level resolution and higher phylogenetic tree node support than when sequences were restricted to the V3-V4 regions (Supplementary Table S1, Fig. S2). However, further direct experimental comparisons using amplicons from the same samples and broadening the number of taxa examined will be necessary to tell whether the advantages we observed for Acinetobacter are generalizable. In addition, shorter read sequencing at higher sequencing depth provides better characterization of rarer bacterial groups which is currently cost prohibitive in general at such depth with the PacBio or other long read sequencing platforms.
Few studies describing the bacterial community in inflow water compared to effluent from a WWTP based on sequence data have been performed despite the potential for contamination of water bodies in highly urbanized areas5,12,13,35,36. Most studies have focused on specific bacterial groups or sampled only inflow water, activated sludge or the effluent. We found distinct compositional differences between the microbiomes of WWTP inflow water and effluent using a whole 16S rRNA gene sequencing approach.
At the phylum level there were two distinct clusters based on inflow and effluent specific bacterial communities, which showed only minor temporal differences. Abiotic parameters such as oxygen concentration as well as competition among different bacterial species with different metabolic characteristics are very likely responsible for the observed differences in bacterial community composition in the WWTP inflow vs. the effluent. At the OTU level, however, there is evidence for seasonal or temporal differences (Table 1), but with only four time points sampled we could not draw any strong conclusions regarding seasonality.
While at the phylum level only minor differences occur between geographically distributed WWTPs, they differ strongly in the composition of the most abundant genera5,12,35,36. For example, our inflow samples shared seven dominant genera with the inflow water of a WWTP in Wisconsin (USA)36 and nine12 or three35 genera with a WWTP in Hong Kong (China). The genera Acinetobacter and Arcobacter were dominant in all studies and are likely common members of WWTPs worldwide5,12,35,36.
The differences could be further explained by other environmental parameters such as pH, temperature and salinity. The WWTP in Hong Kong, for example, treated wastewater has a salinity of 1.2% since it contains ca. 30% seawater used for the toilet flushing system in Hong Kong35. This may possibly favour other bacterial groups in comparison to WWTPs that treat freshwater. Other reasons for the contrasting results might be the use of different small pore size filters for collecting bacteria and the application of different DNA extraction and sequencing methods. While part of the WWTP bacterial community reflects the human microbiome13,61,62, some bacteria likely stem from industrial waste. Environmental bacteria may reach the WWTP via rainfall and wildlife such as rodents inhabiting the drainage system. This might also explain observed regional differences in the bacterial community of WWTPs.
The dominant bacteria found in the current study can be useful or even necessary for the treatment process. Comamonas denitrificans has been shown to be a key organism in WWTPs and thus is very useful by its efficient denitrifying activity63,64. Its higher abundance in the outflow samples agreed with its presence in biofilms of the WWTP facility itself, including activated sludge63,64,65,66. Other species have been identified as abundant members in activated sludge and were suggested to be involved in nutrient removal including nitrite oxidation by Nitrospira spp. or enhanced biological phosphorus removal by Simplicispira limi67,68,69,70,71, which were also abundant in the effluent of the current study.
Bacteria can be harmful for humans and animals by being pathogenic and/or by carrying antibiotic resistance genes. We grouped bacterial genera that contain known pathogenic species into three categories: waterborne, enteric or environmental bacteria that are prone to multidrug resistances. Waterborne bacteria can live in water and use water as vector to spread infection45. Enteric pathogens normally live in the intestines of humans or animals and cause gastrointestinal disease49,50. Transmission of enteric pathogens occurs mainly via the fecal-oral route and contaminated water can serve as a potential vector. Among environmental bacteria are multi-antimicrobial resistant species and species which are potentially pathogenic56,57.
Among waterborne bacteria Vibrio cholera is a well-studied waterborne pathogen46,72 and has been found in WWTPs in Hong Kong, South Africa, USA and Brazil14,73,74,75. Contamination of WWTPs by cholera bacteria is likely human patient derived. As the incidence of cholera in Germany is negligible, this would explain why we never detected OTUs related to the genus Vibrio. Legionella and Leptospira, two other classical waterborne bacterial genera comprise known pathogenic species such as Legionella pneumophila and Leptospira interrogans. The relative abundance of OTUs belonging to these two genera increased from inflow to effluent samples indicating a potential health risk due to contamination of the environment or infection risk for WWTP workers. Legionella spp. are intracellular parasites and can replicate in free-living amoebae76,77. They likely form biofilms in the WWTP, which can promote bacterial growth and persistence in the aquatic environment76,77. In the current study, L. lytica and L. feeleii were identified as closest relatives (Supplementary Fig. S3). While the OTU related to L. lytica is exclusively present in the inflow samples, the OTU related to L. feeleii was detected in both inflow and effluent samples. Both species are known to cause pneumonia in humans when inhaled via aerosols78,79,80,81 and may present a potential health risk as Legionellae in WWTP aerosols are not unusual82,83,84. Wastewater, being enriched in nutrients and carbon, dissolved oxygen concentrations of 6.3–10.3 mg/L, and relatively high temperatures of 14.5–24.6 °C (Supplementary Fig. S10), provides favourable conditions for replication of Legionella spp.85,86,87. These pathogens remain challenging to control as they grow successfully within protozoa and biofilms, where they are relatively protected against disinfectants, grazers and other harsh environmental conditions85.
The increase of Leptospira in the wastewater effluent could be associated with the presence of saprophytic leptospires that reproduce outside of a host and inhabit various aquatic environments88,89. Pathogenic Leptospira, however, can survive in water but do not reproduce outside of a host and thus may be introduced via an infected person or animals such as rodents, which are their natural reservoir and shed leptospires into their environment via urine88,89. Our phylogenetic analyses showed that the OTU affiliated with L. alstonii clustered with known pathogenic species such as L. interrogans and L. mayottensis90,91 and was only present with one read in one of the inflow samples and thus, is likely derived from an infected human or rodent. All other Leptospira OTUs, were exclusively present in the effluent samples and belonged to saprophytic species such as L. idonii and L. biflexa92,93 or were represented by their own cluster (Supplementary Fig. S4). This indicates that wastewater might favour the persistence or possible growth of saprophytic leptospires. While pathogenic leptospires grow much better at temperatures of around 30 °C, saprophytic Leptospira spp. also replicate well at lower temperatures, as low as 10 °C94. The temperatures of our wastewater samples varied between 14.5–24.6 °C during the sampled year (Supplementary Fig. S10). Furthermore, the ability to form biofilms may enhance their survival and/or replication in such an environment. However, as most of these OTUs were related to saprophytic leptospires, we would assume a low health risk potential for humans and animals.
Enteric pathogens can secrete (entero-) toxins, which can damage the gastrointestinal tract of infected individuals95,96,97. They are part of the excreted faecal microbiota of humans in the WWTP inflow, but can also be introduced by animals such as rodents98. In the current study, Clostridium (sensu-stricto), Escherichia-Shigella, and Yersinia were mainly not abundant in the inflow, having a maximum relative abundance of 0.9% and were reduced in or absent from the effluent (Fig. 4). Campylobacter and Salmonella spp. were not detected at all, which could mean that the sequencing depth was too low to detect them. While Escherichia-Shigella and Yersinia decreased in relative abundance, Clostridium (sensu-stricto) remained mainly stable as observed previously12. These findings indicate that the wastewater treatment works well in removing enteric bacteria by introducing oxygen, preventing serious health risk.
Environmental bacteria such as Acinetobacter, Aeromonas and Pseudomonas spp. can be multidrug resistant55,99,100 and some species also have a pathogenic potential such as Pseudomonas aeruginosa and Acinetobacter baumannii101,102. OTUs related to species like P. aeruginosa and A. baumannii were not abundant and only present in the WWTP inflow suggesting that the treatment procedures are effective against these species. Although the overall relative abundance of these three genera was reduced, they were not completely removed during the treatment process.
Pathogens can be strongly diluted in wastewater samples and masked by other much more abundant bacteria. Thus, the presence of pathogens could be greatly underestimated when using 16S rRNA data only. For instance, in a previous study we could detect and isolate C. difficile from the same samples used in the current study and even detect the C. difficile toxin genes via quantitative real-time PCR, but the 16S rRNA dataset did not provide any evidence for the presence of C. difficile103. Therefore, there are clearly limits to high throughput sequencing studies that involve a PCR step in terms of favouring abundant taxa. According to Huse et al.60 sequencing of a hypervariable region covers higher bacterial diversity in comparison to full-length sequencing as a consequence of higher sequencing depth. However, the information provided by full-length 16S rRNA enhances species identification and taxonomic resolution including for potential pathogens. Thus, the choice of the sequencing approach will be based whether less abundant taxa detection or taxonomic resolution are more critical to a given study. In addition, 16S rRNA amplicon data do not reflect absolute abundance of bacteria due to PCR amplification steps and considering the variability in 16S rRNA gene copy numbers among different bacterial taxa104,105. Thus, changes in relative abundances in the current study represent changes in the proportion of bacterial groups between the samples and not absolute quantitative differences. Consequently, an increase in relative abundance (i.e. Legionella or Leptospira) does not necessarily represent an increase in absolute abundance (e.g. growth). The number of bacterial cells in WWTP effluent are usually up to two orders of magnitude lower than in the inflow106,107 This means that an increase of the relative abundance or proportion of Legionella and Leptospira in the effluent does not necessarily reflect an increase in absolute abundance (cell numbers). However, we hypothesize that the efficiency of the wastewater treatment removal capability of such potential pathogenic taxa is lower in comparison to enteric bacteria.
The current study provided evidence for the presence of potential pathogens such as Acinetobacter baumannii, Clostridium perfringens, Legionella lytica, Pseudomonas aeruginosa and Yersinia enterocolitica from the full-length 16S rRNA gene, which may indicate that they are much more abundant than C. difficile, although still rare in the 16S rRNA dataset. Thus, further studies including isolation and cultivation methods are necessary to further investigate the presence and diversity of pathogens, to test for infectivity and to assess a realistic health risk. Water-adapted pathogens, in particularly, such as within the genus Legionella or Leptospira potentially increase in WWTPs and hence should be of great interest for health risk assessment, WWTP operation and waste management.
Material and Methods
Sampling
Untreated raw inflow water and treated effluent of a wastewater treatment plant (WWTP) in Berlin, Germany, were sampled four times in 2016 (February 11th, April 15th, July 27th and October 20th). The sampled effluent had no contact with environment and was already disinfected representing effluent that goes to the environment. The selected WWTP treated municipal wastewater with only a minor percentage of industrial wastewater. It contains a mechanical treatment followed by biological one which includes biological phosphate elimination in combination with nitrification and denitrification, and the production of activated sludge. The effluent undergoes UV sterilization before its release in the environment. The exact location of the sampled WWTP cannot be disclosed due to a confidentiality agreement with the WWTP operators. The water samples were filtered through 0.22 µm Sterivex® filters (EMD Millipore, Germany) connected to a peristaltic pump (EMD Millipore, Germany) to concentrate bacteria and subsequently stored at −20 °C. From the inflow water 20–35 mL could be concentrated on one filter, while from the effluent it was possible to filtrate 175–500 mL. Temperature, pH and dissolved oxygen were measured in the inflow samples with a digital thermometer (Carl Roth, Germany), pH multimeter EC8 (OCS.tec GmbH & CO. KG, Germany), Pen type, IP 67 dissolved oxygen meter (PDO-519, Lutron Electronic Enterprise CO., Taiwan), respectively.
DNA extraction
DNA was extracted from 0.22 µm Sterivex filters using the QIAamp DNA mini kit (Qiagen, Germany) following the protocol for tissue with some modifications. Briefly, the filters were cut into pieces and put into a 2 mL tube. 0.2 µm zirconium glass beads and 360 µL of buffer ATL were added and vortexed for 5 min at 3,000 rpm in an Eppendorf MixMate® (Eppendorf, Germany). Proteinase K (>600 mAU/ml, 40 µL) was added and incubated at 57 °C for 1 h. After centrifugation for 1 min at 11,000 rpm, the supernatant was transferred to a new 2 mL tube and extraction was performed following the manufacturer’s protocol.
Amplification of full-length 16S rRNA genes
Primers 27F (5′-AGRGTTYGATYMTGGCTCAG-3′) and 1492R (5′-RGYTACCTTGTTACGACTT-3′) were used with symmetric barcodes designed by Pacific Biosciences® (USA) for each sample. PCRs for each sample were run in triplicate and carried out in a total volume of 25 µL containing 12.5 µL MyFiTM Mix (Bioline, UK), 9.3 µL water, 0.7 µL of bovine serum albumin (20 mg/mL; New England Biolabs, USA), 0.75 µL of each primer (10 µM) and 1 µL of DNA. The cycling program was as follows: denaturation at 95 °C for 3 min, 25 cycles of 95 °C for 30 sec, 57 °C for 30 sec and 72 °C for 60 sec and a final elongation at 72 °C for 3 min. The quality and concentration of the PCR products were determined using a 4200 TapeStation with D5000 tapes and reagents (Agilent Technologies, USA). Equimolar sample mixes were used for library preparation. Three negative controls were included containing 1 µL water instead of DNA resulting in 13, 13 and 123 total reads representing 5, 8 and 14 OTUs, respectively. OTUs with a read number >5 (=three OTUs in total) in the negative controls were related to the genera Aquabacterium, Ralstonia and Pelomonas, which were subsequently removed from the whole data set prior to analysis.
Library preparation and sequencing
After bead purification with Agencourt AMPure XP (Beckman Coulter, USA), sequencing libraries were built using the SMRTbell Template Prep Kit 1.0‐SPv3 following the guidelines in the amplicon template protocol (Pacific Biosciences, USA). DNA damage repair, end-repair and ligation of hairpin adapters were performed according to the manufacturer’s instruction. DNA template libraries were bound to the Sequel polymerase 2.0 using the Sequel Binding Kit 2.0 (Pacific Biosciences, USA). The data collection per sample was done in a single Sequel SMRT Cell 1M v2 with 600 min movie time on the Sequel system (Pacific Biosciences, USA). We used a 5 pM on-plate loading concentration using Diffusion Loading mode and the Sequel Sequencing Plate 2.0 (Pacific Biosciences, USA).
Sequence analysis
Circular consensus sequences (CCS) for each multiplexed sample were generated with the SMRT Analysis Software (Pacific Biosciences, USA) and used for further downstream analyses. An average of 7 Gb total output per SMRT cell was obtained, with an average CCS read length of 17 kb. Mean amplicon lengths of 1,500 bp were confirmed. For further sequence processing Mothur 1.37 was used108. From a total of 140,092 sequences, 58 sequences with homopolymer stretches of >8 were removed. There were no sequences containing ambiguous bases and further details of the sequencing output such as the average length, error rate and quality are summarized in Supplementary Table S2. Sequences were aligned using the align.seqs command in combination with the Silva v128_SSURef database. Reads that could not be aligned were removed and the remaining sequences were preclustered at 1% difference to account for potential PCR errors and then checked for chimeras using UCHIME in de novo mode109. Classification was done using classify.seqs using the RDP classifier implemented in Mothur110 and Silva v128_SSURef database42,43. Sequences classified as Chloroplast-Mitochondria-unknown-Archaea were removed from the dataset. Operational taxonomic unit (OTU) clustering was done with VSEARCH (dgc mode;111) as implemented in Mothur, using a 99% similarity cutoff to nearly represent one species per OTU. This cutoff was used to resolve relationships among closely related bacteria that would be masked when using a cutoff of 97%. Phylogenetic analyses were performed with the ARB software using the LTPs128_SSU tree112 and the SILVA database for bacterial 16S rRNA genes42,43.
Phylogenetic analyses and statistics
Maximum-likelihood phylogenies (PhyML) were built with Jukes Cantor as the substitution model including 1,000 bootstrap replicates by using Geneious® 9.0.5113. To compare full-length with short read sequences, we restricted the sequences affiliated with the genus Acinetobacter using 16S rRNA primers to a 464 bp amplicon covering the hypervariable regions 3-441. Beta-diversity analyses (i.e. box plots, PCoA and bar charts) were performed after rarefaction and log standardization of OTU-counts table using R version 3.5. and the package vegan. The differential abundance of OTUs in the inflow versus the effluent was computed on the non-rarefied OTU counts. The test used the exact negative binomial test in combination with the quantile-adjusted conditional maximum likelihood estimation of dispersion of the R package edgeR114. This analysis was based on TMM (trimmed mean of M values, where M is the log-fold-change of each OTU) normalized abundance data115. The test basically performed a pairwise comparison of OTU relative abundances between the two sample groups and an OTU was considered to respond significantly when the Bonferroni-corrected p-value was below 0.01.
Data Availability
The dataset generated and analysed during the current study are available from the Sequence Read Archive (SRA) of NCBI (National Center for Biotechnology Information) under the BioProject ID PRJNA484334 and SRA accession SRP156296.
References
D’Arcy, B. & Frost, A. The role of best management practices in alleviating water quality problems associated with diffuse pollution. Sci. Total Environ. 265, 359–367 (2001).
McLellan, S. L., Fisher, J. C. & Newton, R. J. The microbiome of urban waters. Int. Microbiol. Off. J. Span. Soc. Microbiol 18, 141–149 (2015).
Niemczynowicz, J. Urban hydrology and water management – present and future challenges. Urban Water 1, 1–14 (1999).
Wang, J., Da, L., Song, K. & Li, B.-L. Temporal variations of surface water quality in urban, suburban and rural areas during rapid urbanization in Shanghai, China. Environ. Pollut. 152, 387–393 (2008).
Marti, E., Jofre, J. & Balcazar, J. L. Prevalence of Antibiotic resistance genes and bacterial community composition in a river influenced by a wastewater treatment plant. PLOS ONE 8, e78906 (2013).
Miao, X.-S., Bishay, F., Chen, M. & Metcalfe, C. D. Occurrence of antimicrobials in the final effluents of wastewater treatment plants in Canada. Environ. Sci. Technol. 38, 3533–3541 (2004).
Michael, I. et al. Urban wastewater treatment plants as hotspots for the release of antibiotics in the environment: A review. Water Res 47, 957–995 (2013).
Munir, M., Wong, K. & Xagoraraki, I. Release of antibiotic resistant bacteria and genes in the effluent and biosolids of five wastewater utilities in Michigan. Water Res 45, 681–693 (2011).
Rizzo, L. et al. Urban wastewater treatment plants as hotspots for antibiotic resistant bacteria and genes spread into the environment: a review. Sci. Total Environ. 447, 345–360 (2013).
Szczepanowski, R. et al. Detection of 140 clinically relevant antibiotic-resistance genes in the plasmid metagenome of wastewater treatment plant bacteria showing reduced susceptibility to selected antibiotics. Microbiol. Read. Engl 155, 2306–2319 (2009).
Vajda, A. M. et al. Demasculinization of male fish by wastewater treatment plant effluent. Aquat. Toxicol. 103, 213–221 (2011).
Cai, L., Ju, F. & Zhang, T. Tracking human sewage microbiome in a municipal wastewater treatment plant. Appl. Microbiol. Biotechnol. 98, 3317–3326 (2014).
Newton, R. J. et al. Sewage reflects the microbiomes of human populations. mBio 6, e02574–14 (2015).
Cai, L. & Zhang, T. Detecting human bacterial pathogens in wastewater treatment plants by a high-throughput shotgun sequencing technique. Environ. Sci. Technol. 47, 5433–5441 (2013).
Wakelin, S. A., Colloff, M. J. & Kookana, R. S. Effect of wastewater treatment plant effluent on microbial function and community structure in the sediment of a freshwater stream with variable seasonal flow. Appl. Environ. Microbiol. 74, 2659–2668 (2008).
Ferguson, A. S. et al. Comparison of fecal indicators with pathogenic bacteria and rotavirus in groundwater. Sci. Total Environ. 431, 314–322 (2012).
Vaz-Moreira, I., Nunes, O. C. & Manaia, C. M. Bacterial diversity and antibiotic resistance in water habitats: searching the links with the human microbiome. FEMS Microbiol. Rev. 38, 761–778 (2014).
Bennett, P. M. Plasmid encoded antibiotic resistance: acquisition and transfer of antibiotic resistance genes in bacteria. Br. J. Pharmacol 153, S347–S357 (2008).
Roe, E., Jones, R. J. & Lowbury, E. J. L. Transfer of antibiotic resistance between Pseudomonas aeruginosa, Escherichia coli, and other Gram-negative bacilli in burns. Lancet 149–52 (1971).
Courvalin, P. Transfer of antibiotic resistance genes between gram-positive and gram-negative bacteria. Antimicrob. Agents Chemother. 38, 1447–1451 (1994).
Zhang, Y., Marrs, C. F., Simon, C. & Xi, C. Wastewater treatment contributes to selective increase of antibiotic resistance among Acinetobacter spp. Sci. Total Environ. 407, 3702–3706 (2009).
Bonomo, R. A. & Szabo, D. Mechanisms of multidrug resistance in Acinetobacter species and Pseudomonas aeruginosa. Clin. Infect. Dis 43, S49–S56 (2006).
Kaskhedikar, M. & Chhabra, D. Multiple drug resistance in Aeromonas hydrophila isolates of fish. Food Microbiol 28, 157–168 (2010).
Trautmann, M., Lepper, P. M. & Haller, M. Ecology of Pseudomonas aeruginosa in the intensive care unit and the evolving role of water outlets as a reservoir of the organism. Am. J. Infect. Control 33, S41–S49 (2005).
Silge, A. et al. Identification of water-conditioned Pseudomonas aeruginosa by Raman microspectroscopy on a single cell level. Syst. Appl. Microbiol. 37, 360–367 (2014).
Selezska, K. et al. Pseudomonas aeruginosa population structure revisited under environmental focus: impact of water quality and phage pressure. Environ. Microbiol 14, 1952–1967 (2012).
Kersters, I. et al. Survival potential of Aeromonas hydrophila in freshwaters and nutrient-poor waters in comparison with other bacteria. J. Appl. Bacteriol 80, 266–276 (1996).
Warburton, D. W., McCormick, J. K. & Bowen, B. Survival and recovery of Aeromonas hydrophila in water: development of methodology for testing bottled water in Canada. Can. J. Microbiol. 40, 145–148 (1994).
Brandi, G., Sisti, M., Giardini, F., Schiavano, G. F. & Albano, A. Survival ability of cytotoxic strains of motile Aeromonas spp. in different types of water. Lett. Appl. Microbiol. 29, 211–215 (1999).
Cateau, E., Verdon, J., Fernandez, B., Hechard, Y. & Rodier, M.-H. Acanthamoeba sp. promotes the survival and growth of Acinetobacter baumanii. FEMS Microbiol. Lett 319, 19–25 (2011).
Karumathil, D. P., Yin, H.-B., Kollanoor-Johny, A. & Venkitanarayanan, K. Effect of chlorine exposure on the survival and antibiotic gene expression of multidrug resistant Acinetobacter baumannii in. Water. Int. J. Environ. Res. Public. Health 11, 1844–1854 (2014).
Warskow, A. L. & Juni, E. Nutritional requirements of Acinetobacter strains isolated from soil, water, and sewage. J. Bacteriol. 112, 1014–1016 (1972).
Kooij, D., van der, Visser, A. & Hijnen, Wa. M. Growth of Aeromonas hydrophila at Low Concentrations of Substrates Added to Tap Water. Appl. Environ. Microbiol. 39, 1198–1204 (1980).
Sautour, M., Mary, P., Chihib, N. E. & Hornez, J. P. The effects of temperature, water activity and pH on the growth of Aeromonas hydrophila and on its subsequent survival in microcosm water. J. Appl. Microbiol . 95, 807–813 (2003).
Ye, L. & Zhang, T. Bacterial communities in different sections of a municipal wastewater treatment plant revealed by 16S rDNA 454 pyrosequencing. Appl. Microbiol. Biotechnol. 97, 2681–2690 (2013).
McLellan, S. L., Huse, S. M., Mueller-Spitz, S. R., Andreishcheva, E. N. & Sogin, M. L. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ. Microbiol 12, 378–392 (2010).
Logares, R., Mangot, J.-F. & Massana, R. Rarity in aquatic microbes: placing protists on the map. Res. Microbiol. 166, 831–841 (2015).
Logares, R. et al. Patterns of rare and abundant marine microbial eukaryotes. Curr. Biol. 24, 813–821 (2014).
Saunders, A. M., Albertsen, M., Vollertsen, J. & Nielsen, P. H. The activated sludge ecosystem contains a core community of abundant organisms. ISME J. 10, 11–20 (2016).
Pedrós-Alió, C. The rare bacterial biosphere. Annu. Rev. Mar. Sci 4, 449–466 (2012).
Pruesse, E., Peplies, J. & Glöckner, F. O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1–e1 (2013).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–D596 (2013).
Yilmaz, P. et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res 42, D643–D648 (2014).
Hunter, P. R., Colford, J. M., LeChevallier, M. W., Binder, S. & Berger, P. S. Waterborne diseases. Emerg. Infect. Dis. 7, 544 (2001).
Colwell, R. R. Infectious disease and environment: cholera as a paradigm for waterborne disease. Int. Microbiol. 7, 285–289 (2004).
Cameron, D. & Jones, I. G. John Snow, the Broad Street Pump and Modern Epidemiology. Int. J. Epidemiol. 12, 393–396 (1983).
Cann, K. F., Thomas, D. R., Salmon, R. L., Wyn-Jones, A. P. & Kay, D. Extreme water-related weather events and waterborne disease. Epidemiol. Infect. 141, 671–686 (2013).
Ketley, J. M. Pathogenesis of enteric infection by Campylobacter. Microbiology 143, 5–21 (1997).
Bartlett, J. G. Clostridium difficile: history of its role as an enteric pathogen and the current state of knowledge about the organism. Clin. Infect. Dis 18, S265–S272 (1994).
Smedley, J. G., Fisher, D. J., Sayeed, S., Chakrabarti, G. & McClane, B. A. The enteric toxins of Clostridium perfringens. in Reviews of Physiology, Biochemistry and Pharmacology 183–204, https://doi.org/10.1007/s10254-004-0036-2 (Springer Berlin Heidelberg, 2005).
Broz, P., Ohlson, M. B. & Monack, D. M. Innate immune response to Salmonella typhimurium, a model enteric pathogen. Gut Microbes 3, 62–70 (2012).
Wren, B. W. The Yersiniae — a model genus to study the rapid evolution of bacterial pathogens. Nat. Rev. Microbiol. 1, 55–64 (2003).
Formal, S. B., Hale, T. L. & Sansonetti, P. J. Invasive enteric pathogens. Rev. Infect. Dis 5, S702–S707 (1983).
Figueira, V., Vaz-Moreira, I., Silva, M. & Manaia, C. M. Diversity and antibiotic resistance of Aeromonas spp. in drinking and waste water treatment plants. Water Res 45, 5599–5611 (2011).
Khan, N. H. et al. Isolation of Pseudomonas aeruginosa from open ocean and comparison with freshwater, clinical, and animal isolates. Microb. Ecol. 53, 173–186 (2007).
Guardabassi, L., Dalsgaard, A. & Olsen, J. E. Phenotypic characterization and antibiotic resistance of Acinetobacter spp. isolated from aquatic sources. J. Appl. Microbiol. 87, 659–667 (1999).
Holmes, P., Niccolls, L. M. & Sartory, D. P. The ecology of mesophilic Aeromonas in the aquatic environment. Genus Aeromonas 127–150 (1996).
Park, R. W. A. A Study of certain heterotrophic polarly flagellate water bacteria: Aeromonas, Pseudomonas and Comamonas. Microbiology 27, 121–133 (1962).
Huse, S. M. et al. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLOS Genet. 4, e1000255 (2008).
Fisher, J. C. et al. Comparison of sewage and animal fecal microbiomes by using oligotyping reveals potential human fecal indicators in multiple taxonomic groups. Appl. Environ. Microbiol. 81, 7023–7033 (2015).
Huttenhower, C. et al. Structure, function and diversity of the healthy human microbiome. Nature 486, 207 (2012).
Andersson, S., Dalhammar, G., Land, C. J. & Rajarao, G. K. Characterization of extracellular polymeric substances from denitrifying organism Comamonas denitrificans. Appl. Microbiol. Biotechnol. 82, 535 (2009).
Gumaelius, L., Magnusson, G., Pettersson, B. & Dalhammar, G. Comamonas denitrificans sp. nov., an efficient denitrifying bacterium isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 51, 999–1006 (2001).
Andersson, S. Characterization of bacterial biofilms for wastewater treatment. DIVA (2009).
Andersson, S., Kuttuva Rajarao, G., Land, C. J. & Dalhammar, G. Biofilm formation and interactions of bacterial strains found in wastewater treatment systems. FEMS Microbiol. Lett 283, 83–90 (2008).
Alawi, M., Off, S., Kaya, M. & Spieck, E. Temperature influences the population structure of nitrite-oxidizing bacteria in activated sludge. Environ. Microbiol. Rep 1, 184–190 (2009).
Lemaire, R., Yuan, Z., Blackall, L. L. & Crocetti, G. R. Microbial distribution of Accumulibacter spp. and Competibacter spp. in aerobic granules from a labscale biological nutrient removal system. Environ. Microbiol. 10, 354–363 (2008).
Lücker, S. et al. Nitrotoga-like bacteria are previously unrecognized key nitrite oxidizers in full-scale wastewater treatment plants. ISME J. 9, 708–720 (2015).
You, S. J., Hsu, C. L. & Ouyang, C. F. Identification of the microbial diversity of wastewater nutrient removal processes using molecular biotechnology. Biotechnol. Lett 24, 1361–1366 (2002).
Lu, S. et al. Simplicispira limi sp. nov., isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 57, 31–34 (2007).
Swerdlow, D. L. et al. Waterborne transmission of epidemic cholera in Trujillo, Peru: lessons for a continent at risk. The Lancet 340, 28–32 (1992).
Barrett, T. J. et al. Use of Moore swabs for isolating Vibrio cholerae from sewage. J. Clin. Microbiol. 11, 385–388 (1980).
Levine, M. M. et al. The pathogenicity of nonenterotoxigenic Vibrio cholerae serogroup 01 Biotype EI Tor isolated from sewage water in Brazil. J. Infect. Dis. 145, 296–299 (1982).
Okoh, A. I. & Igbinosa, E. O. Antibiotic susceptibility profiles of some Vibrio strains isolated from wastewater final effluents in a rural community of the Eastern Cape Province of South Africa. BMC Microbiol. 10, 143 (2010).
Bartram, J., Chartier, Y., Lee, J. V., Pond, K. & Surman-Lee, S. Legionella and the prevention of legionellosis. (World Health Organization, 2007).
Declerck, P. et al. Replication of Legionella pneumophila in biofilms of water distribution pipes. Microbiol. Res. 164, 593–603 (2009).
Lee, J., Caplivski, D., Wu, M. & Huprikar, S. Pneumonia due to Legionella feeleii: case report and review of the literature. Transpl. Infect. Dis 11, 337–340 (2009).
Marrie, T. J., Raoult, D., La Scola, B., Birtles, R. J. & de Carolis, E. Legionella-like and other amoebal pathogens as agents of community-acquired pneumonia. Emerg. Infect. Dis. 7, 1026–1029 (2001).
McNally, C., Hackman, B., Fields, B. S. & Plouffe, J. F. Potential importance of Legionella species as etiologies in community acquired pneumonia (CAP). Diagn. Microbiol. Infect. Dis. 38, 79–82 (2000).
Muder, R. R. & Victor, L. Y. Infection due to Legionella species other than L. pneumophila. Clin. Infect. Dis. 35, 990–998 (2002).
Medema, G., Wullings, B., Roeleveld, P. & Kooij, Dvander Risk assessment of Legionella and enteric pathogens in sewage treatment works. Water Sci. Technol. Water Supply 4, 125–132 (2004).
Pascual, L. et al. Bioaerosol emission from wastewater treatment plants. Aerobiologia 19, 261–270 (2003).
Roll, B. M. & Fujioka, R. S. Detection of legionella bacteria in sewage by polymerase chain reaction and standard culture method. Water Sci. Technol 31, 409–416 (1995).
Molofsky, A. B. & Swanson, M. S. Differentiate to thrive: lessons from the Legionella pneumophila life cycle. Mol. Microbiol 53, 29–40 (2004).
Wullings, B. A. & van der Kooij, D. Occurrence and genetic diversity of uncultured Legionella spp. in drinking water treated at temperatures below 15 °C. Appl. Environ. Microbiol. 72, 157–166 (2006).
Söderberg, M. A. & Cianciotto, N. P. Mediators of lipid A modification, RNA degradation, and central intermediary metabolism facilitate the growth of Legionella pneumophila at low temperatures. Curr. Microbiol. 60, 59–65 (2010).
Aviat, F. et al. Leptospira exposure in the human environment in France: A survey in feral rodents and in fresh water. Comp. Immunol. Microbiol. Infect. Dis. 32, 463–476 (2009).
Wynwood, S. J. et al. Leptospirosis from water sources. Pathog. Glob. Health 108, 334–338 (2014).
Bourhy, P., Collet, L., Brisse, S. & Picardeau, M. Leptospira mayottensis sp. nov., a pathogenic species of the genus Leptospira isolated from humans. Int. J. Syst. Evol. Microbiol. 64, 4061–4067 (2014).
Ren, S.-X. et al. Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature 422, 888–893 (2003).
Saito, M. et al. Leptospira idonii sp. nov., isolated from environmental water. Int. J. Syst. Evol. Microbiol. 63, 2457–2462 (2013).
Picardeau, M. et al. Genome sequence of the saprophyte Leptospira biflexa provides insights into the evolution of Leptospira and the pathogenesis of leptospirosis. PLOS ONE 3, e1607 (2008).
Johnson, R. C. & Harris, V. G. Differentiation of pathogenic and saprophytic leptospires I. Growth at low temperatures. J. Bacteriol. 94, 27–31 (1967).
Burgen, A. S. V., Dickens, F. & Zatman, L. J. The action of botulinum toxin on the neuro‐muscular junction. J. Physiol 109, 10–24 (1949).
Delor, I. & Cornelis, G. R. Role of Yersinia enterocolitica Yst toxin in experimental infection of young rabbits. Infect. Immun. 60, 4269–4277 (1992).
Wassenaar, T. M. Toxin production by Campylobacter spp. Clin. Microbiol. Rev. 10, 466–476 (1997).
Arnone, R. D. & Walling, J. P. Waterborne pathogens in urban watersheds. J. Water Health 5, 149–162 (2007).
van Dessel, H. et al. Identification of a new geographically widespread multiresistant Acinetobacter baumannii clone from European hospitals. Res. Microbiol. 155, 105–112 (2004).
Odjadjare, E. E. et al. Prevalence of multiple antibiotics resistant (MAR) Pseudomonas species in the final effluents of three municipal wastewater treatment facilities in South Africa. Int. J. Environ. Res. Public. Health 9, 2092–2107 (2012).
Mena, K. D. & Gerba, C. P. Risk assessment of Pseudomonas aeruginosa in water. in. Reviews of Environmental Contamination and Toxicology 201, 71–115 (2009).
Peleg, A. Y., Seifert, H. & Paterson, D. L. Acinetobacter baumannii: Emergence of a successful pathogen. Clin. Microbiol. Rev. 21, 538–582 (2008).
Numberger, D. et al. Genomic analysis of three Clostridioides difficile isolates from urban water sources. Anaerobe, https://doi.org/10.1016/j.anaerobe.2019.01.002 (2019).
Piwosz, K. et al. Determining lineage-specific bacterial growth curves with a novel approach based on amplicon reads normalization using internal standard (ARNIS). ISME J. 12, 2640–2654 (2018).
Větrovský, T. & Baldrian, P. The Variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLOS ONE 8, e57923 (2013).
Timraz, K., Xiong, Y., Al Qarni, H. & Hong, P.-Y. Removal of bacterial cells, antibiotic resistance genes and integrase genes by on-site hospital wastewater treatment plants: surveillance of treated hospital effluent quality. Environ. Sci. Water Res. Technol 3, 293–303 (2017).
Ma, L. et al. Rapid quantification of bacteria and viruses in influent, settled water, activated sludge and effluent from a wastewater treatment plant using flow cytometry. Water Sci. Technol. J. Int. Assoc. Water Pollut. Res. 68, 1763–1769 (2013).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Ludwig, W. et al. ARB: a software environment for sequence data. Nucleic Acids Res 32, 1363–1371 (2004).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol 59, 307–321 (2010).
Robinson, M. D. & Smyth, G. K. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9, 321–332 (2008).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Acknowledgements
We thank Claudia Quedenau for technical assistance to produce sequencing data, Daniele Y. Sunaga-Franze for computational support as well as the wastewater treatment plant in Berlin, which allowed us to do the sampling. The IGB-MIBI group is thanked for their discussions and technical support. This work was supported by a project grant (IRG 3 - Water) from the Leibniz Research Alliance “INFECTIONS’21 - Transmission Control of Infections in the 21st Century” as funded by the Leibniz Association, Germany (SAS-2015-FZB-LFV) and the Leibniz SAW project “MycoLink” (SAW-2014-IGB).
Author information
Authors and Affiliations
Contributions
The study was designed by D.N., H.P.G. and A.D.G. D.N. collected and performed most of the practical work as well as data analysis and manuscript writing. L.G. processed the sequence data, L.Z. contributed by testing for differential abundance, K.M. supported the study with important scientific discussion, and S.S. provided the PacBio® Sequel sequencing expertise and sequencing data. All authors commented on the manuscript, read and agreed with the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Numberger, D., Ganzert, L., Zoccarato, L. et al. Characterization of bacterial communities in wastewater with enhanced taxonomic resolution by full-length 16S rRNA sequencing. Sci Rep 9, 9673 (2019). https://doi.org/10.1038/s41598-019-46015-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-46015-z
This article is cited by
-
Ecological features of trace elements tolerant microbes isolated from sewage sludge of urban wastewater treatment plant
Stress Biology (2024)
-
Microbial communities and their roles in the Cenozoic sulfurous oil reservoirs in the Southwestern Qaidam Basin, Western China
Scientific Reports (2023)
-
Enhanced Anaerobic Mono-digestion and Co-digestion of Crop Residues by NaOH Alkali Pre-treatment: Digestion Performance and Microbial Community Dynamics
Waste and Biomass Valorization (2023)
-
Spatial and Seasonal Variations in the Bacterial Community of an Anthropogenic Impacted Urban Stream
Microbial Ecology (2023)
-
Stronger Geographic Limitations Shape a Rapid Turnover and Potentially Highly Connected Network of Core Bacteria on Microplastics
Microbial Ecology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
