
Abstract
Chikungunya virus (CHIKV) is considered a public health problem due to its rapid spread and high morbidity. This study aimed to determine the genetic diversity and phylogenetic relationships of CHIKVs in Colombia. A descriptive and retrospective study was carried out using sera of patients infected with Chikungunya during the outbreak in Colombia. The whole genomes of CHIKV (n = 16) were sequenced with an Illumina Hi-seq 2500 and were assembled using the Iterative Virus Assembler software. A Bayesian inference phylogenetic analysis was carried out with 157 strains of worldwide origin. The Colombian CHIKV sequences were grouped in the Asian genotype; however, three independent phylogenetic subclades were observed, probably the result of three separate introductions from Panama, Nicaragua, and St. Barts. Each subclade showed several different non-synonymous mutations (nsP2-A153V; nsp2-Y543H; nsp2-G720A; nsP3-L458P; Capside R78Q), that may have functional consequences for CHIKV biology and pathogenesis. These same mutations may affect the efficacy of potential CHIKV vaccines.
Similar content being viewed by others

Introduction
Chikungunya virus (CHIKV) is an alphavirus of the Togaviridae family, that is transmitted by the bite of mosquitoes of the species Aedes aegypti and A. albopictus1. CHIKV is considered a public health problem due to its rapid spread and high morbidity2. It causes a febrile illness accompanied by maculopapular rash and severe joint pain that can last for months or years1. The incapacitating nature of the disease has caused a substantial economic burden and collapse in health systems1. The local transmission of CHIKV has been reported in more than 100 countries and territories in Asia, Africa, Europe, and the Americas1. According to the World Health Organization (WHO), between December 2013, and 2017, the islands of the Caribbean and the Americas registered >2,5 million suspected and confirmed3.
The study of whole genome sequences of CHIKV isolates from different regions of the world has facilitated an understanding of the evolutionary history of this virus4. The first phylogenetic studies of CHIKV identified three geographically associated genotypes: West Africa (WA), East/Central/South Africa (ECSA) and Asia (AS)5. However, in the years 2005–2006 a new descendant from the ECSA, the Indian Ocean lineage (IOL) was described6. IOL developed a higher affinity for the Aedes albopictus vector, resulting from the E1-A226V, E2-I211T, E1-T98A and E2-L210Q mutations2, and was associated with the massive epidemics of 2005–2006 in the Indian Ocean Islands and the Indian subcontinent6,7. Since the outbreaks in India and the Indian Ocean, CHIKV has spread worldwide, and the locations of epidemics have changed drastically8,9. In 2007, the first outbreak of transmission in Europe was reported in Italy and in 2013, the first cases in the Caribbean island of Saint Martin10. In January 2015 CHIKV was disseminated to 42 countries or territories in the Caribbean, Latin America and North America (Florida)11,12.
In recent decades, Colombia has suffered epidemics due to a large number of emerging arboviruses such as dengue virus, Venezuelan equine encephalitis virus, CHIKV, and Zika, which have caused severe public health problems13. The first case of local transmission of CHIKV in Colombia was notified in August 2014 in the department of Bolivar14. According to the Pan American Health Organization, Colombia was the country with the third highest number of cases after Brazil and the Dominican Republic15; between 2014 and 2015 there were 460,484 cases diagnosed by clinical symptoms and 4,658 confirmed by the laboratory16,17. Finally, 21,149 cases have been notified between 2016 and August 2018, demonstrating the endemicity of Chikungunya in Colombia18.
The rapid distribution of CHIKV could be related to the mobility of people and merchandise. According to the National Institute of Health (INS), in Colombia, the first reported cases were imported from Venezuela, the Dominican Republic and Panama19. Previous phylogenetic analyses with partial sequences of genes nsP1, E1, and E2 in Colombia, found that CHIKV was similar to the Asian genotype and was related to isolates from Saint Martin Island, Virgin Islands, Mexico and Brazil14,20. However, these phylogenetic studies did not analyze the complete genome or sequences of many countries affected by CHIKV, which precluded a comprehensive evaluation of the genetic variability, and phylogenetic relationships of the strains studied21.
Previous studies have determined that nucleotide substitution rates occur more frequently during urban transmission than in enzootic transmission21. It is known that the Asian genotype circulates mainly in an urban transmission cycle and has a genomic evolution rate of 4.71 (95% BCI: 3.84–5.65) nucleotide substitutions per year, higher than the genotype. ECSA (2.31, 95% BCI: 1.89 to 2.71)21,22. There is evidence that some Asian strains in the Colombian Caribbean are already exhibiting nucleotide substitutions in proteins nsP1 and E2 20, but it is unknown if there are variations in other regions of the viral genome that are associated with autochthonous vectors or the clinical manifestations of patients.
The present study aimed to determine the genetic diversity, phylogenetic relationships, and possible routes of introduction of CHIKV strains isolated in Colombia and the Americas.
Results
Viral isolation
CHIKV was isolated from 96.5% (54/57) of the sera, 65.4% (n = 36/54) of the isolated viruses coming from Cartagena, 29% (n = 16/54) from Ovejas, 1.8% from Planeta Rica and 1.8% from San Joaquín-Mahates (SJ-Mahates). Positive cultures were obtained from samples with viral loads of 1.19 × 102 and 1, 25 × 107 viruses/mL, which generally correspond to the first four days of the acute phase of the disease. In the three sera from which viral isolation was not achieved, the viral loads were less than 4.5 × 101 and corresponded to the fifth day of disease. The three strains donated by the Colombian National Institute of Health (INS) were isolated in C6/36 and Vero cells. The most marked difference between the Vero cell line and C6/36 was the cell lysis and the time of appearance of the cytopathic effect (CPE). It was observed that the CPE in C6/36 cells occurred from the fifth day with the formation of syncytia. The C6/36 cell monolayer remained stable for approximately seven days. In contrast, in the Vero cells, the CPE was observed at 24–48 hours post-infection (hpi) and was characterized by plaque formation (24 hpi) and cell lysis between 3–4 days.
Genome structure and genetic variability of CHIKV isolated in Colombia
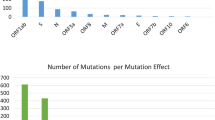
For each sequence, an average of 35,574,564 paired reads (32,021,178 and 40,904,884) and 3,602,990,602 read bases were obtained. The percentage of GC was 49%. The 16 genomes were assembled with an average length of 12062 (11,394 to 12,347) nucleotides. The average percentage value of the coding regions of the genome (ORF) of CHIKV was 28.8% adenine (A), 25.9% guanine (G), 20.6% thymine (T) and 24.7% cytosine (C). As reported for other CHIKVs, the sequences contained two open reading frames of 7410 nt and 3744 nt that encode the nonstructural polyprotein (2470 aa nsP1-nsP4) and the structural polyprotein (1248 aa E1-E3, 6 K, C), respectively. The length of genes of the strains and the percentage of homology concerns to reference genome LN898093 (Supplementary Table S1). Sixty polymorphic sites were detected among the Colombian sequences (Fig. 1). The 70% (n = 42) of the substitutions occurred in the non-structural proteins and 30% (n = 18) in the structural proteins. We found 8 polymorphic sites in Nsp1 (6 ti + 2 tv), 13 in Nsp2 (11 ti + 2tv), 9 in Nsp3 (6ti + 3tv), 12 in Nsp4 (10ti + 2 tv), 4 in capsid (4 ts), 4 in E2 (4ts), 1 in 6 K (1ts) and 9 in E1 (6 ts + 3 tv). Sixteen out of the 61 polymorphisms were non-synonymous substitutions, and of these, 4 were parsimoniously informative, and 12 were non-informative (Fig. 1).

Number of mutations detected in the strains of CHIKV isolated in the present study. Each gene has a range nucleotides position depicted below the genes nsP1 to E1. Each gene shows polymorphic sites (yellow), transitions (blue), transversions (red) and nonsynonymous mutation (green). The Colombian sequences previously published in the Gen-Bank were not taken into account for this analysis.
Molecular fingerprint and phylogenetic relationship of Colombian CHIKV
Within the Caribbean American clade (CAC), the Colombian strains were grouped into three subclades with strains of different geographical origins. Each subclade had branch support of value 1 (posterior probability 1), indicating a genuine relationship between grouped strains. i. the Panamanian strain (KR559486-2014) was close to those of the Colombian Caribbean (SJ-Mahates, Cartagena, Ovejas, Planeta Rica, Sincelejo) and Huila; ii. The Nicaraguan strain (KY703969-2014) was close to Risaralda and Cauca; and iii. St. Barts strain (KR559497-2014) was closely related to two Colombian strains, one reported in Bogota and the other one of unknown origin (Fig. 2 and Supplementary Fig. S2).

Bayesian inference tree. Taxon labels include access number, country of isolation and year of collection. Subsequent probabilities ≥0.80 are labeled in each branch.
After phylogenetic analysis, polymorphic sites that allowed subclade grouping were found into the strains from America and the Caribbean Islands (Fig. 3). It was observed that the strains of CAC had 8 polymorphic sites; those modifications were different from the ancestral Asiatic strains (Fig. 3). Within the CAC, some polymorphisms allowed differentiation into sub-clades from different countries (Fig. 3). In the case of the Colombian strains and phylogenetically close ones (Nicaragua, Saint Barts, Panamá), it was observed that the mutations T3308C (aa. nsp2-Y543H), G3840C (aa. nsp2-G720A) and T5445C (aa. nsP3-L458P) were only present in the strains of Nicaragua, Risaralda, and Cauca. The mutations nsP3-A5356G, nsP4-C6676T, and capsid-G7787A (aa. capsid-R78Q) were present in 3 isolates from St. Barts, Bogotá and unknown Colombian strain with no geographic description in GenBank. The nsP3-G4324T mutation appeared in the strains of the Dominican Republic, USA (Florida and California), Puerto Rico, Honduras, Venezuela, Panama, the Colombian Caribbean, and Huila. Additionally, T2139C (aa. nsP2-A 153 V) and nsP2-C2305T mutations were detected in the strains of Panama (KR559486), Colombian Caribbean and Huila (Figs 3, 4; Table 1).

Nucleotide changes that defined subclades within the Caribbean American clade. The gray color represents the nucleotides similar to the Philippine strain AB860301. The yellow color represents a change to guanine, red to cytosine, blue to thymine and green to adenine. Black represents mixed bases within the sequences.

Chikungunya virus spread in the Americas based on the phylogenetic tree and molecular fingerprint. Different colors represent several mutations in Caribbean American clades of CHIKV. Yellow color shows the islands where the initial strains were introduced in the Americas from the Philippines. Pink color shows strains with mutations nsP3-A5356G, nsP4-C6676T and capsid-G7787A (aa. capsid-R78Q); green color (nsP4-G5949A [aa. nsP4-R 99Q]); Orange color (nsP3-G4324T); Orange color with lines (nsP3-G4324T, nsP2-C2305T, nsP2-T2139C [aa. nsP2-A153V]); Blue color (T479C, C1525T); Purple color (nsP2-T3308C [aa. nsp2-Y543H], nsP2-G3840C [aa. nsp2-G720A] and nsP3-T5445C [aa. nsP3-L458P]); green apple color: (nsP4-T6931A, E3-A8490G).
When we compared the sequences with the closest phylogenetic strain, 40 variable sites were detected between the Panamanian strain (KR559486) and those from SJ-Mahates, Ovejas, Cartagena, Planeta Rica, Sincelejo and Huila (Supplementary Table S3). Of the 40 sites, 37 were unique mutations, and 11 produced changes in the amino acid sequence (Table 1). 72% (n = 29/40) of the substitutions occurred in the non-structural proteins and 28% (n = 11/40) in the structural proteins. We found 5 polymorphic sites in nsP1 (4 ti + 1 tv), 8 in nsP2 (7 ti + 1tv), 7 in nsP3 (5ti + 2tv), 9 in nsP4 (7ti + 2 tv), 4 in capsid (4 ts), 3 in E2 (3ts), and 4 in E1 (3 ts + 1 tv) (Supplementary Table S3). The strains with the most variable sites were those from the Planeta Rica (Córdoba) and Huila, with 12 and 9 polymorphisms, respectively.
When comparing the strains of Risaralda and Cauca with that of Nicaragua (KY703969), 18 unique mutations were detected, one of which produced a change in the amino acid sequence (Table 1 and Supplementary Table S3). 50% (n = 9/18) of the substitutions occurred in the non-structural proteins and 50% (n = 9/18) in the structural proteins. We found 3 polymorphic sites in nsP1 (2 ti + 1 tv), 2 in nsP2 (2ts), 4 in nsP4 (4 ts), 2 in E2 (2ts), 1 in 6 K (1 ts) and 6 in E1 (4 ts + 2 tv) (Supplementary Table S3). The strains of St. Barts and Colombia contained two polymorphisms (2ts + 2tv) of which one produced a change in the amino acid sequence (Table 1 and Supplementary Table S3).
Discussion
The phylogenetic analysis in this study determined that CHIKVs in Colombia belong to 3 clusters of the Asiatic genotype: Panama (Caribbean Colombia, Huila), Nicaragua (Cauca and Risaralda) and St. Barts (Bogotá, D.C). Our results are similar to previous research that used complete sequencing of worldwide strains and a Bayesian phylogenomics approach7,23. In this sense, the previous analysis including three Colombian viral genomes, two from Sincelejo (Sucre) (KU365372.1-KU365373.1) and one from Bogotá D.C, identified a similar phylogenetic relationship between Panama and St. Barts, respectively7,23. However, that work focused on a global phylogenic study, and few Colombian strains were analyzed.
On the other hand, our results are different from other studies on phylogenic analyses of Colombian CHIKV. Mattar et al.20 in 2015 and Rodas et al.24 in 2016, evaluated Colombian Caribbean strains (Sincelejo, Ovejas, SJ-Mahates) with partial sequences of the E2, 6 K, E1 and nsP1 genes and found a phylogenetic relationship with strains from Virgin Islands, Saint Lucia, Mexico, Puerto Rico, Brazil and China20,24. Laiton-Donato et al.14 in 2015 used the E1 gene to study isolates from 15 departments in Colombia and found that there was a relationship with CHIKV from Mexico, Brazil, Virgin Islands, and Saint Martin14. Other recent studies25 in Colombia evaluated strains of Norte de Santander and found a relationship with the Colombian Caribbean strains and China25. It should be noted that the previous phylogenetic analyses carried out in Colombia included strains of few countries and partial sequences of genes, thus precluding a complete evaluation of dynamic evolution and phylogenetic relationships of studied strains21. In contrast, our research used whole viral genomes and 157 isolates from affected countries, allowing us to identify a significant number of informative sites, reflecting a virus evolutionary history.
When analyzing the polymorphisms, the mutations nsP2-C-2305-T and nsP2-C-2139 and (AA, nsP2-V153A) confirmed the phylogenetic relationship between the isolates of Panama, the Colombian Caribbean, and Huila (Figs 3, 4). The mutation nsP3 G-4324-T suggested that the strains of Panama, Colombian Caribbean, and Huila could come from Venezuela, the United States, Puerto Rico, the Dominican Republic or Honduras (Figs 3, 4). Of the countries that make up this clade, the Dominican Republic was the first country to report cases of indigenous transmission of CHIKV (February 2014)26 followed by Puerto Rico (May 2014)3, USA, Venezuela, Brazil, Panama (July 2014)3,22 and Colombia (September 2014)3,27. With the information systems of migratory passenger mobility and the surveillance system in Public Health of Colombia, it was possible to establish that during May and July 2014, infected travelers from the Dominican Republic arrived in Panama and Colombia respectively (Epidemiological Bulletin Colombia)19,28. This epidemiological information suggests that the route of introduction to the Colombian Caribbean was through a strain that was circulating previously on the Dominican Republic and later was introduced to Panamá, Colombian Caribbean, and Huila.
On May 13, 2014, Panama reported its first imported cases passengers coming from the Dominican Republic and Haiti29. Colombia’s first imported cases came from Venezuela, the Dominican Republic, and Panama and were reported more than two months later, in late July19. The high migration of travelers between Panama and Colombia facilitates the transmission of diseases between these countries; according to the Ministry of Foreign Affairs of Colombia, in July 2014, there were 34,413 travelers between Panama and Colombia28. For the same period, Cartagena was the Colombian city with the third highest number of foreigners (20,800 people)28. Therefore, it is possible that the Panamanian strain of CHIKV has entered through Cartagena (Bolivar) and not through SJ-Mahates (50 km away from Cartagena), where the first autochthonous cases were detected27.
In interviews carried out with the habitants of SJ-Mahates and its surrounding rural areas, it was established that infected people from Venezuela and Cartagena preceded the time of the outbreak in SJ-Mahates27. The first cases in Cartagena were related to those cases arriving from Venezuela and the Dominican Republic27. This epidemiological information also allows us to suggest that the introduction of CHIKV to the Colombian Caribbean may have come from Venezuela and the Dominican Republic and the additional mutations nsP2 C-2305-T, T-2139-C (aa.ns P2-V153A) arose in Colombia and were imported into Panama.
In the last 5 to 10 years, Venezuela has faced a severe economic crisis, precipitated by political instability30. Public health has been affected, and the country is experiencing an increase and expansion of vector-borne diseases30. The United Nations High Commissioner for Refugees estimates that between 2014 and March 2018, around 600,000 people sought refuge in Colombia, not counting the people who migrated through illegal border crossings30. Despite the migration of Venezuelans to Colombia and all the cases of CHIKV that were imported from Venezuela to Colombia before and during the epidemic27, in this study no close phylogenetic relationship was found with Venezuelan strains. However, the circulation of these strains in Colombian border areas with Venezuela, such as Guajira and Norte de Santander cannot be ruled out. In this sense, the possibility for detecting Venezuelan or other countries’ strains in Colombia was affected by the limited number of sequences from some territories of the Americas and the absence of samples from other areas of Colombia.
Concerning the strains found in the Risaralda and Cauca Andean area, the T3308C (aa. nsp2-Y543H), G3840C (aa. nsp2-G720A) and T5445C (aa. nsP3-L458P) mutations demonstrate their phylogenetic relationship with the Nicaraguan strain (Figs 3, 4). In a previous study, it was determined that the Nicaraguan strain was closely related to the strain of British Virgin Islands31. In contrast, our study showed that strain BVI KJ451624 has a relationship to Mexican strain (Fig. 2). BVI KJ451624 strain was one of the first to appear in the outbreak in January 2014 in the Caribbean islands; it is possible that the strains of CHIKV mutated in their movement from Sant Martin December 2013, British Virgin Islands, Mexico, Nicaragua, and Colombia, where the mutations were detected.
It is difficult to determine how the Nicaraguan strain reached the South West of the Andean region of the country (Risaralda and Cauca). One possibility is that the strain entered from Nicaragua through the Island of San Andres since the border is only 233 km away and San Andres is one of the most visited tourist sites by people of the Andean area of Colombia (Fig. 4). However, it could also be in the opposite direction, so that the strains of San Andres islands migrated to Nicaragua. Unfortunately, in this study, it was not possible to include strains of San Andres thus not allowing us to make inferences about this phenomenon of viral circulation.
Regarding the strain Saint Barts 2014, the phylogenetic analysis demonstrated a close relationship with two Colombian strains of 2014 (KR559491) and 2016 (KX496989) (Figs 2, 3). The strain KX496989 was isolated from an American woman who traveled to Bogotá D.C and had a co-infection with Zika virus and CHIKV32. The altitude of Bogotá at 2600 meters above sea level, does not support the populations of the Aedes aegypti vector33; however, is possible that the woman was infected in another municipality of Cundinamarca, since in the description of the clinical case it is indicated that the woman visited urban and rural areas where she received mosquito bites32. According to the reports of the National Institute of Health, between the years 2014–2016 in Cundinamarca, there were 19,919 cases of CHIKV17,18. In this sense, our study suggests that the CHIKV of Saint Barts circulates in Cundinamarca or nearby departments.
The detection of polymorphisms A5356G, C6676T and G7787A (aa.C-R78Q) in strains isolated more than two years apart (2014 to 2016), suggests that the CHIKV of Saint Bart and Colombia is a variant within the Caribbean American clade (Fig. 3). Interestingly, the G7787A mutation (aa Capside-R78Q) was also detected in the car-89 strain of Cartagena (Table 1). This finding in strain Car-89 may have been the result of an error-matched viral RNA polymerase, or due to a recombination event, which has already been reported for CHIKV and other alphaviruses. In this study, we have no certainty of what could have happened, and neither were analyzes performed to detect possible recombinant events. It would be interesting to investigate if this variant still circulates in Colombia and if additional mutations have occurred from 2016 to 2019.
In addition to the mutations that were identified in each clade, we found that the Colombian strains had 60 polymorphic sites, of which 70% (n = 42/60) occurred in non-structural proteins and 30% (n = 18) in structural proteins (Table 1 and supplementary Table S3). These findings are similar to those reported by Stapleford et al.34, who through deep sequencing searched for the frequency of viral minority populations in the same individual and found that the majority of CHIKV polymorphisms occurred in non-structural proteins and 3′UTR34.
The biological function of synonymous and non-synonymous substitutions in Colombian CHIKV is unknown, and it would be interesting to carry out future studies to understand how these changes in the genome influence the pathogenesis and transmission of CHIKV35,36. Of the adaptive mutations in E1 and E2 previously reported, the Colombian strains contain the mutations E2-G60D and E2-I211T37. It has been proved that E2-G60D increases the infectivity in A. albopictus and A. aegypti, whatever the amino acid (alanine or valine) in position E1-2263636. On the other hand, E2-I211T increases the infectivity in Ae. albopictus of strains CHIKV E1-226V, but does not affect Ae. Aegypti37,38. It is important to note that Colombian strains, as well as other Asian strains, contain threonine in position 98 of E1 (E1-98T), which limits their ability to infect Ae. albopictus through the substitution E1-A226V37.
In conclusion, the genetic diversity and polymorphism in several American countries of CHIKV genomes implied evolutionary diversification and possible adaptation to human ecosystems and insect vectors. The subclades showed different non-synonymous mutations with likely functional consequences for CHIKV biology and pathogenesis.
Methods
Ethical approval and informed consent
Institutional standard guidelines of the Minister of Health of Colombia and the University of Cordoba ethics committee were followed for the collection of patients’ blood samples after their written informed consent for involvement in the study was obtained. Sera of pediatric patients from Hospital Infantil Napoleon Franco, Cartagena, Colombia were authorized by the ethics committee of the Hospital (Acta # 6 October 15th, 2015) and informed consent was obtained from all patients under the authorization of their parents. All patients included in the study were under an anonymous code number. The study incorporated procedures, management, and conservation of samples, and technical-administrative procedures for health research required by resolution 8430 of the Ministry of Health of Colombia, in 199339, and declaration of Helsinki for ethical and medical research in human subjects40.
Specimens
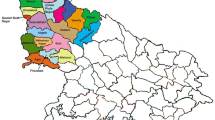
Between September and December 2014 during the epidemic outbreak in the Caribbean area of Colombia, a collection of 57 sera was selected20,41. Thirty-six sera were from Cartagena (Department of Bolivar), 19 from Ovejas (Department of Sucre), 1 from San Joaquin-Mahates (Department of Bolivar) and 1 from Planeta Rica (Department of Córdoba). The specimens came from pediatric and adult patients, had complete clinical and epidemiological records and were positive for CHIKV by qRT-PCR (LightMix Chikungunya Virus Kit, Roche, USA). The viral loads ranged between 1.06 × 101–1.25 × 107 copies/mL. The National Institute of Health of Colombia (INS) donated three strains isolated from the Departments of Risaralda, Huila, and Cauca, all of them belonging to the Andean area of Colombia (Fig. 5).

Map of Colombia showing the isolation sites of the strains studied.
Cell culture and viral isolation
The 57 sera were cultured onto C6/36 and Vero cells, maintained in 12.5 cm2 flasks, in Leibovitz 15 and DMEM (Dulbecco’s Modified Eagle Medium) media respectively. Media were supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 1X non-essential amino acids, 100 units/ml penicillin, 100 µg/ml streptomycin and 2 µg amphotericin B. C6/36 cells were incubated at 28 °C and Vero cells 37 °C under 5% CO2. For viral isolation, 40 µl of serum were added to confluent monolayers of C6/36 or Vero cells and incubated for 1 hour at 28 °C and 37 °C respectively. After viral adsorption, the sample was removed, and 3 ml of medium supplemented with 2% FBS was added and incubated at 28 °C or 37 °C according to the cell line. At the end of 14 day or following early recognition of the cytopathic effect (CPE), the cellular supernatants were collected and stored at −80 °C. All the cell culture procedures were carried out at Biological Research Institute of the Tropic of the University of Cordoba with equipment’s and efficient practices of a level of biosafety II, that involve infectious or potentially infectious material.
RNA extraction, RT-PCR, and confirmation of CHIKV
The RNA was obtained from 140 μl of the supernatant of the cell cultures using the QIAamp Viral RNA Mini Kit (Qiagen, France), following the manufacturer’s recommendations. For the synthesis of cDNA, M-MLV Reverse Transcriptase (200 U/μl, Promega) and Random Primers (0.5 μg/μl, Promega) were used. For the confirmation of CHIKV in viral cultures, TaqMan Fast Universal PCR Master Mix (2X) (Applied Biosystems) was used. For each reaction, 10 µl of 2X buffer were added; 0.18 µl of forward primer (final concentration 0.9 µM); 0.18 µl of reverse primer (final concentration 0.9 μM); 0.1 µl of probe (final concentration 0.25 μM); 4.54 µl of RNase-free water and 5 µl of cDNA. As positive controls, a culture of CHIKV confirmed by conventional PCR was used; ultra-pure water served as a negative control. Samples with a Ct less than or equal to 38 were considered positive
Amplification of the CHIKV genome by PCR
16 viral isolates of CHIKV were selected based on their geographical origin: Cartagena n = 7, San Joaquin-Mahates = 1, (Department of Bolivar), Ovejas n = 4 (Department of Sucre), Planeta Rica n = 1 (Department of Cordoba), Department of Risaralda n = 1, Department of Cauca n = 1, Department of Huila n = 1 (Table 2). For amplification of the CHIKV genome, twelve sets of primers previously reported by Stapleford et al.34 with modifications, were used (Supplementary Table S4). The primers amplified overlapping regions of the retrotranscribed genomes. The PCR reaction was carried out with the Exact Taq PLUS 2X Mastermix. The amplified products were verified by electrophoresis in 1% agarose gel using SYBR safe DNA Gel Stain. The amplicons of each sample were mixed in equimolar concentration, to avoid unequal distribution of data in the steps of library preparation and sequencing.
Sequencing and assembly of genomes
The libraries were prepared using a TruSeq DNA Sample Prep Kit (Illumina). Generation of clusters and fragment sequencing was performed using a Truseq SBS Kit v4 and Illumina HiSeq 2500 (2-x 101pb paired-end). The fastq files were processed using Trimmomatic v0.3642, for the removal of low-quality sequences. The genomes were assembled with the Iterative Virus Assembler software and the reference strain LN898093 of Martinique43.
Preparation of data
A first dataset (CHIKV3gen; n = 173) was generated with the 16 sequences assembled in this study and 157 complete genomes of CHIKV available in GenBank. The GenBank sequences belonged to the three genotypes, with collection dates between the years 1953–2016 (Supplementary Table S5). The 173 sequences were aligned with MUSCLE v3.8.3144 and manually curated while retaining the codon homology with the Geneious v11.1.2 program45. A second dataset (CHIKVas; n = 104) was prepared from CHIKV3gen and contained only Asian CHIKVs from 1953–2016. Due to the ambiguous alignment of the UTR, only the open reading frames (ORFs) comprising 11256 nucleotides were used for the reconstruction of the phylogenetic tree and subsequent analyses. The nucleotide and amino acid positions used were based on the reference sequences KX262995, KY055011 and LN898093. All sequences were identified by the date of sampling and the location from which they originated.
Phylogenetic reconstruction
The Bayesian phylogenetic inference of the ORF alignments was made using a molecular and uncorrelated clock with the BEAST v2 program46. The analysis was run in duplicate with 90 million replicates, and the trees were sampled every 1000 steps. The coalescence model implemented was a Bayesian Skyline prior with a relaxed clock model and variation rates between branches using a log-normal distribution and the substitution model recommended by jModel Test (GTR + G + I). The convergence of MCMC chains was verified with Tracer v1. 7. The trees obtained were edited using the software FigTree v1.4.3. The alignment of the second dataset CHIKVas; (n = 104) which contained Asian genotypes was used to determine the genetic diversity (Software MEGA 7).
References
An, W., Ge, N., Cao, Y., Sun, J. & Jin, X. Recent progress on chikungunya virus research. Virologica Sinica 32, 441–453, https://doi.org/10.1007/s12250-017-4072-x (2017).
Tsetsarkin, K. A., Chen, R. & Weaver, S. C. Interspecies transmission and chikungunya virus emergence. Current opinion in virology 16, 143–150, https://doi.org/10.1016/j.coviro.2016.02.007 (2016).
Pan American Health Organization, World Health Organization. Number of reported cases of Chikungunya fever in the Americas: Epidemiological Week 1–52, 2014, https://www.paho.org/hq/index.php?option = com_topics&view = rdmore&cid = 7928&item = chikungunya&cat = statistics&type = 2014-7928&Itemid = 40931&lang = en (2014).
de Lamballerie, X. et al. Chikungunya virus adapts to tiger mosquito via evolutionary convergence: a sign of things to come? Virology journal 5, 33, https://doi.org/10.1186/1743-422X-5-33 (2008).
Abraham, R. et al. Correlation of phylogenetic clade diversification and in vitro infectivity differences among Cosmopolitan genotype strains of Chikungunya virus. Infection, genetics and evolution 37, 174–184, https://doi.org/10.1016/j.meegid.2015.11.019 (2016).
Shragai, T., Tesla, B., Murdock, C. & Harrington, L. C. Zika and chikungunya: mosquito-borne viruses in a changing world. Annals of the New York Academy of Sciences 1399, 61–77, https://doi.org/10.1111/nyas.13306 (2017).
Sahadeo, N. S. D. et al. Understanding the evolution and spread of chikungunya virus in the Americas using complete genome sequences. Virus evolution 3, vex010, https://doi.org/10.1093/ve/vex010 (2017).
Chen, M. W. et al. Chikungunya virus nsP4 RNA-dependent RNA polymerase core domain displays detergent-sensitive primer extension and terminal adenylyltransferase activities. Antiviral research 143, 38–47, https://doi.org/10.1016/j.antiviral.2017.04.001 (2017).
Schwartz, O. & Albert, M. L. Biology and pathogenesis of chikungunya virus. Nature reviews. Microbiology 8, 491–500, https://doi.org/10.1038/nrmicro2368 (2010).
Yang, S., Fink, D., Hulse, A. & Pratt, R. D. Regulatory considerations in development of vaccines to prevent disease caused by Chikungunya virus. Vaccine 35, 4851–4858, https://doi.org/10.1016/j.vaccine.2017.07.065 (2017).
Vu, D. M., Jungkind, D. & Angelle Desiree, L. Chikungunya Virus. Clinics in laboratory medicine 37, 371–382, https://doi.org/10.1016/j.cll.2017.01.008 (2017).
Weber, C. et al. Identification of Functional Determinants in the Chikungunya Virus E2 Protein. PLoS neglected tropical diseases 11, e0005318, https://doi.org/10.1371/journal.pntd.0005318 (2017).
Mendez, N., Oviedo-Pastrana, M., Mattar, S., Caicedo-Castro, I. & Arrieta, G. Zika virus disease, microcephaly and Guillain-Barre syndrome in Colombia: epidemiological situation during 21 months of the Zika virus outbreak, 2015–2017. Archives of public health=Archives belges de sante publique 75, 65, https://doi.org/10.1186/s13690-017-0233-5 (2017).
Laiton-Donato, K. et al. Phylogenetic analysis of Chikungunya virus in Colombia: Evidence of purifying selection in the E1 gene. Biomedica: revista del Instituto Nacional de Salud 36, 25–34, https://doi.org/10.7705/biomedica.v36i0.2990 (2015).
Pan American Healt Organization. Geographic Spread of Chikungunya in the Americas for december 2013–December 2017. http://ais.paho.org/phip/viz/ed_chikungunya_amro.asp (2017).
Instituto Nacional de Salud. Comportamientos de los eventos de vigilancia en salud pública. Boletines epidemiológico 53 de 2014. https://www.ins.gov.co/buscador-eventos/BoletinEpidemiologico/2014%20Boletin%20epidemiologico%20semana%2053.pdf (2014).
Instituto Nacional de Salud. Comportamientos de los eventos de vigilancia en salud pública. Boletin epidemiológico 52 de 2015. https://www.ins.gov.co/buscador-eventos/BoletinEpidemiologico/2015%20Boletin%20epidemiologico%20Semana%2052.pdf (2015).
Instituto Nacional de Salud. Comportamientos de los eventos de vigilancia en salud pública. Boletin epidemiológico 52 de 2016; Boletin epidemiológico 52 de 2017; Boletin epidemiológico 17 de 2018. https://www.ins.gov.co/buscador-eventos/Paginas/Vista-Boletin-Epidemilogico.aspx (2014).
Instituto Nacional de Salud. Comportamientos de los eventos de vigilancia en salud pública. Boletin epidemiológico 37 de 2014. https://www.ins.gov.co/buscador-eventos/BoletinEpidemiologico/2014%20Boletin%20epidemiologico%20semana%2037.pdf (2014).
Mattar, S. et al. Outbreak of Chikungunya virus in the north Caribbean area of Colombia: clinical presentation and phylogenetic analysis. Journal of infection in developing countries 9, 1126–1132, https://doi.org/10.3855/jidc.6670 (2015).
Volk, S. M. et al. Genome-scale phylogenetic analyses of chikungunya virus reveal independent emergences of recent epidemics and various evolutionary rates. Journal of virology 84, 6497–6504, https://doi.org/10.1128/JVI.01603-09 (2010).
Nunes, M. R. et al. Emergence and potential for spread of Chikungunya virus in Brazil. BMC medicine 13, 102, https://doi.org/10.1186/s12916-015-0348-x (2015).
Chen, R. et al. Comprehensive Genome Scale Phylogenetic Study Provides New Insights on the Global Expansion of Chikungunya Virus. Journal of virology 90, 10600–10611, https://doi.org/10.1128/JVI.01166-16 (2016).
Rodas, J. D. et al. Genetic Characterization of Northwestern Colombian Chikungunya Virus Strains from the 2014-2015 Epidemic. The American journal of tropical medicine and hygiene 95, 639–646, https://doi.org/10.4269/ajtmh.16-0091 (2016).
Carrillo-Hernandez, M. Y., Ruiz-Saenz, J., Villamizar, L. J., Gomez-Rangel, S. Y. & Martinez-Gutierrez, M. Co-circulation and simultaneous co-infection of dengue, chikungunya, and zika viruses in patients with febrile syndrome at the Colombian-Venezuelan border. BMC infectious diseases 18, 61, https://doi.org/10.1186/s12879-018-2976-1 (2018).
Pimentel, R., Skewes-Ramm, R. & Moya, J. Chikungunya en la República Dominicana: lecciones aprendidas en los primeros seis meses. Rev Panam Salud Publica 36, 336–341 (2014).
Instituto Nacional de Salud. Informe Final del Evento Chikungunya, Colombia 2014. http://www.ins.gov.co/buscador-eventos/Informesdeevento/Chikungun%CC%83a%202014.pdf (2014).
Ministerio de Relaciones Exteriores. Boletin migratorio Julio del 2014. http://migracioncolombia.gov.co/index.php/es/?option = com_content&view = article&id = 718 (2014).
Carrera, J. P. et al. Unusual pattern of chikungunya virus epidemic in the Americas, the Panamanian experience. PLoS neglected tropical diseases 11, e0005338, https://doi.org/10.1371/journal.pntd.0005338 (2017).
Grillet, M. E. et al. Venezuela’s humanitarian crisis, resurgence of vector-borne diseases, and implications for spillover in the region. The Lancet Infectious Diseases, https://doi.org/10.1016/s1473-3099(18)30757-6 (2019).
Wang, C. et al. Chikungunya Virus Sequences Across the First Epidemic in Nicaragua, 2014–2015. The American journal of tropical medicine and hygiene 94, 400–403, https://doi.org/10.4269/ajtmh.15-0497 (2016).
Cherabuddi, K. et al. Zika and Chikungunya virus co-infection in a traveller returning from Colombia, 2016: virus isolation and genetic analysis. JMM case reports 3 (2016).
Ruiz-López, F. et al. Presence of Aedes (Stegomyia) aegypti (Linnaeus, 1762) and its natural infection with dengue virus at unrecorded heights in Colombia. Biomedica: revista del Instituto Nacional de Salud 36, 303–308 (2016).
Stapleford, K. A. et al. Whole-Genome Sequencing Analysis from the Chikungunya Virus Caribbean Outbreak Reveals Novel Evolutionary Genomic Elements. PLoS neglected tropical diseases 10, e0004402, https://doi.org/10.1371/journal.pntd.0004402 (2016).
Hawman, D. W. et al. Mutations in the E2 Glycoprotein and the 3′ Untranslated Region Enhance Chikungunya Virus Virulence in Mice. Journal of virology, https://doi.org/10.1128/JVI.00816-17 (2017).
Tsetsarkin, K. A. et al. Epistatic roles of E2 glycoprotein mutations in adaption of chikungunya virus to Aedes albopictus and Ae. aegypti mosquitoes. PloS one 4, e6835, https://doi.org/10.1371/journal.pone.0006835 (2009).
Zouache, K. & Failloux, A.-B. Insect–pathogen interactions: contribution of viral adaptation to the emergence of vector-borne diseases, the example of chikungunya. Current Opinion in Insect Science 10, 14–21, https://doi.org/10.1016/j.cois.2015.04.010 (2015).
Tsetsarkin, K. A., Chen, R., Sherman, M. B. & Weaver, S. C. Chikungunya virus: evolution and genetic determinants of emergence. Current opinion in virology 1, 310–317, https://doi.org/10.1016/j.coviro.2011.07.004 (2011).
Republica de Colombia, Ministerio de Salud, Resolucion N° 008430 DE 1993. Por la cual se establecen las normas científicas, técnicas y administrativas para la investigación en salud (4 de octubre de 1993).
WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects. https://www.wma.net/wp-content/uploads/2016/11/DoH-Oct2013-JAMA.pdf (2013).
Paternina-Caicedo, A. et al. Features of Dengue and Chikungunya Infections of Colombian Children under 24 Months of Age Admitted to the Emergency Department. Journal of tropical pediatrics 64, 31–37, https://doi.org/10.1093/tropej/fmx024 (2018).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120, https://doi.org/10.1093/bioinformatics/btu170 (2014).
Hunt, M. et al. IVA: accurate de novo assembly of RNA virus genomes. Bioinformatics 31, 2374–2376, https://doi.org/10.1093/bioinformatics/btv120 (2015).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC bioinformatics 5, 113, https://doi.org/10.1186/1471-2105-5-113 (2004).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649, https://doi.org/10.1093/bioinformatics/bts199 (2012).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus evolution 4, vey016, https://doi.org/10.1093/ve/vey016 (2018).
Acknowledgements
To Colciencias project number 111271250545. To the University of Córdoba, Colombia, Vice-rectoria of Investigation, sustainability program for researching groups, 2016–2017 (Code FMV-01-16 and FMV-01-17). To National Institute of Health from Colombia (INS) for the donation of the strains. To Professor Ben Adler, for the grammar and style corrections.
Author information
Authors and Affiliations
Contributions
Conceived and designed study S.M., Y.V.W.; laboratory tests: Y.V.W., G.A., S.M.; analyzed data: Y.V.W., A.P., S.M., C.M., G.A., R.H.; wrote the paper: Y.V.W., S.M., A.P., C.M., R.H., D.P., G.A., H.P.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Villero-Wolf, Y., Mattar, S., Puerta-González, A. et al. Genomic epidemiology of Chikungunya virus in Colombia reveals genetic variability of strains and multiple geographic introductions in outbreak, 2014. Sci Rep 9, 9970 (2019). https://doi.org/10.1038/s41598-019-45981-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-45981-8
This article is cited by
-
Chikungunya Encephalitis: an Inconsistently Reported Headache and Cause of Death in Patients with Pre-Existing Conditions
Current Tropical Medicine Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
