
Abstract
Chronic neuroinflammation is known to contributes to the toxicity of neurodegeneration of Parkinson’s disease (PD). However, the molecular and cellular mechanisms controlling inflammatory responses in the central nervous system remain poorly understood. Here we found that a E3 ubiquitin ligase Peli1 is dramatically induced only in the substantia nigra (SN) of the human and mouse PD brains. The ablation of Peli1 significantly suppressed LPS-induced production of neurotoxic mediators and proinflammatory cytokines in SN and in primary microglia, whereas Peli1 is dispensable for the inflammatory responses in astrocyte. Accordingly, Peli1 deficiency markedly inhibited neuron death induced by the conditioned medium from LPS-stimulated microglia. Mechanistical study suggested that Peli1 acts as a positive regulator of inflammatory response in microglia through activation of NF-κB and MAP kinase. Our results established Peli1 as a critical mediator in the regulation of microglial activation and neuroinflammation-induced death of dopaminergic neurons during PD pathogenesis, suggesting that targeting Peli1 may have therapeutic effect in neuroinflammation.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is a neurodegenerative disease among people over 65 years old that causes the movement dysfunction, which is due to the death of tyrosine hydroxylase (TH)+ dopaminergic neurons in the substantia nigra (SN)1,2. Published studies have suggested that microglia-associated neuroinflammation is a common feature during PD pathogenesis3,4,5. Microglia are the innate immune cells that reside very early in the central nervous system (CNS), in which they surveil the local environment to maintain the CNS homeostasis under physiological condition. Under pathological conditions, microglia are over-activated and produce large amounts of neurotoxic mediators, such as interleukin (IL)-1β, and tumor necrosis factor (TNF), nitric oxide (NO)5,6. During PD pathogenesis, rather than exhibiting protective function, activated microglia are detrimental for the survival of dopaminergic neurons and contribute to the aggregation of disease progression6,7,8,9.
Microbial infections have long been recognized as an environmental trigger for the pathogenesis of PD10. Lipopolysaccharide (LPS), endotoxin from gram-negative bacteria, is a potent activator of microglia and damages dopaminergic neurons only in the presence of microglia11,12. In addition, intracranial infusion of LPS could induce the death of TH+ dopaminergic neurons in mice, and thus was commonly applied for the generation of animal models to mimic the PD symptoms13. Microglia were found to be the first responder and rapidly activated in LPS-induced inflammation, producing large amounts of neurotoxic factors. These factors like IL-1β can activate astrocyte to produce more neurotoxic mediators together with that produced by microglia, leading to the loss of dopaminergic neurons14. These studies collectively suggested that bacteria infection may be a critical mediator to promote the disease onset and progression of PD.
LPS is a ligand for toll-like receptor (TLR)4, which induces MyD88- and Trif-mediated activation of downstream signals, such as NF-κB, MAP kinases and interferon regulatory factors (IRFs), leading to the transcriptional activation of neurotoxic genes like Il1b, Tnf, Nos2, etc15. The Pellino (Peli) E3 ubiquitin ligases have been reported to play essential roles in the modulation of TLR, IL-1 receptor (IL-1R) and TNR receptor (TNRR) signaling in innate immune cells16,17,18,19, and in the regulation of adaptive immune cell activation20,21. Our recent studies have indicated that Peli1 protein is highly expressed in the brain and spinal cord, in which Peli1 is predominantly expressed in microglia, but not in other CNS-resident cells like neuron, astrocytes and oligodendrocytes. In such case, Peli1 functions as an important mediator for microglia activation and contribute to the pathogenesis of multiple sclerosis and viral encephalitis19,22.
Based on ours and others’ previous reports, the current study hypothesized that Peli1 could regulate the PD pathogenesis through modulating microglial inflammatory responses. Our experiments were first aimed to characterize Peli1 function in PD and secondly to decipher the mechanism how Peli1 regulate the survival of dopaminergic neurons during neuroinflammation.
Results
Peli1 is induced in the SN of human and mouse PD brain
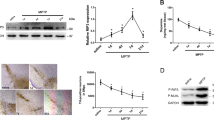
Recent evidences have suggested that not only CNS-resident cells, but also peripheral immune system, critically regulate the pathology of PD23,24. Since we previously demonstrated that Peli1 plays important roles in regulating the function of both CNS-resident microglia and peripheral immune cells19,20,22, so in order to examine the biological function of PELI1 during PD pathogenesis, we initially searched the public NCBI GEO database (GDS2519, GDS2821, GDS3128, GDS3129 and GDS4145) and examined the relative expression of PELI1 in SN, medullary regions, and peripheral blood mononuclear cells (PBMCs) that isolated from health donors (HD) and PD patients. We found that not only one dataset (GDS2821, GDS3128 and GDS3129) suggested that PELI1 expression is dramatically increased in the SN of PD patients as compared to that of HD (P = 0.0196, P = 0.0008, and P = 0.0451 Fig. 1a). Although there is no statistical difference, the data from GDS4145 suggested that the PELI1 expression levels have an increased tendency in the medullary regions of PD patients as compared to that from HD (Fig. 1a). However, there is no obvious difference of PELI1 mRNA expression in PBMCs between HD and PD patients (Fig. 1b). To confirm these observations, we stereotaxically injected LPS into the mouse SN to generate the inflammation-induced PD model, and examine the Peli1 induction. The result revealed that Peli1 mRNA and its protein expression in SN is dramatically increased upon LPS priming as compared to that in PBS-injected control SN (Fig. 1c,d). These data collectively suggested that Peli1 may function directly in CNS-resident cells but not in the peripheral immune system to regulate PD pathogenesis.

Peli1 is induced in the substantia nigra of human and mouse PD brains. (a,b) Normalized PELI1 expression signals in peripheral blood mononuclear cells (PBMCs) (a) and substantia nigra (SN) (b) from health donors (HD) and Parkinson’s disease (PD) patients, the data were obtained from NCBI GEO dataset (GDS2821, GDS3128, GDS3129, GDS4145, GDS2519). Each dot in the graphs represent a value from one person. (c,d) QPCR and immunoblot analysis of relative mRNA (c) and protein (d) expression for Peli1 in the SN of WT and Peli1-KO mice that stereotaxically injected with 2 μl PBS or LPS (5 μg/ml). The immunoblot results are presented as Peli1 and Actin blot panels (d, left panel) and summary bar graph quantifying the relative Peli1 protein levels to Actin (d, right panel). Data with error bars represent mean ± SD. Each panel is representative of three independent experiment. *P < 0.05, ***P < 0.001 as determined by unpaired Student’s t test.
Peli1 deficiency protects against inflammation-induced TH+ neurons death
To figure out whether Peli1 indeed modulate PD pathogenesis, we generated the PD model by injecting LPS into SN of WT and Peli1-KO mice, and then analyzed the survival condition of TH+ dopaminergic neurons by using the immunohistochemical staining of SN 7 days after LPS treatment. The data suggested that LPS injection induced a remarkable death of TH+ neurons in SN as reported. In contrast, Peli1 deficiency markedly suppressed the loss and enhanced the survival rate of TH+ neurons in SN as compared to that in WT mice (Fig. 2a,b). Accordingly, we observed there are more accumulation of Iba1+ microglia and GFAP+ astrocytes in the SN of WT PD mice than that in KO PD mice (Fig. 2c,d), suggesting the suppressed activation of the glial cells in Peli1-KO mice during PD pathogenesis.

Peli1 deficiency suppresses inflammation-induced neuron death in substantia nigra. WT and Peli1-deficient mice (n = 4) were stereotaxically injected with 2 μl LPS (5 μg/ml) or equal volume of PBS into the substantia nigra (SN) of each mouse. (a,b) Immunohistochemical analysis of tyrosine hydroxylase (TH) in the ventral mesencephalon sections taken 7 days after PBS or LPS injection in WT and Peli1-KO mice. Data are showed as representative images (a) and summary bar graph showing the survival rates by calculating the ratios of TH+ neuron numbers of LPS-treated SN as compared to that of PBS-treated SN of the same mouse (b). (c,d) Immunofluorescent staining for Iba1 to indicate microglia, and GFAP to indicate astrocyte on the ventral mesencephalon of WT and Peli1-KO mice that injected with PBS or LPS. Data were presented as representative images (c) and summary bar graph showing the relative integrated optical density (IOD) of Iba1 and GFAP reactivity (d). Data with error bars represent mean ± SD. Each panel is representative of at least three independent experiment. *P < 0.05 as determined by unpaired Student’s t test (b) or two-way ANOVA with a Bonferroni post test (d).
Peli1 contributes to the enhanced inflammation in SN of PD mice
To confirm that Peli1 mediates the enhanced inflammation in SN during PD pathogenesis, we examined the expression of proinflammatory genes in SN (Fig. 3a). In consistent with the immunofluorescence data, Peli1 deficiency markedly inhibited LPS-induced expression of Nos2 and proinflammatory genes (Il1b, Il6, Tnf) in SN as compared to that injected with PBS (Fig. 3b), suggesting that Peli1 served as a positive regulator of neuroinflammation in PD pathology.

Peli1 deficiency suppresses LPS-induced neuroinflammation in substantia nigra. (a) Experimental diagram showing the procedure of LPS-induced neuroinflammation in SN and the time points when to collect tissue sample for QPCR or histological analysis. (b) QPCR analysis of relative mRNA expression for Nos2 and the indicated proinflammatory genes in the SN of WT and Peli1-KO mice that stereotaxically injected with 2 μl PBS or LPS (5 μg/ml). The gene expression were normalized to a reference gene Actb (encoding β-actin) and showed as the relative value to Actb in bar graphs. Data with error bars represent mean ± SD. Each panel is representative of at least three independent experiment. **P < 0.01 as determined by two-way ANOVA with a Bonferroni post test (b).
Peli1 mediates LPS-induce production of neurotoxic factors in microglia
It is reported that microglia are initially activated and responsible for the neurotoxicity of LPS-mediated neuroinflammation in SN25, so we tested the LPS-mediated gene induction in mouse primary microglia. Expectedly, we found that LPS-induced expression of Nos2 and proinflammatory genes that encoding IL-1β, IL-6 and TNF, were significantly inhibited in Peli1-deficient microglia (Fig. 4a). Accordingly, the loss of Peli1 in microglia significantly suppressed the secretion of these proinflammatory cytokines and neurotoxic nitrites in the microglial cell culture supernatant (Fig. 4b). Therefore, these results suggested that Peli1 mediates the activation and production of neurotoxic factors in microglia during PD pathogenesis.

Peli1 mediates LPS-induce inflammatory responses in microglia. (a) QPCR analysis of relative mRNA expression for the indicated inflammatory genes in the primary cultured microglia isolated from WT and Peli1-KO mice, left untreated (NT) or stimulated with LPS (100 ng/ml). (b) ELISA of IL-1β, IL-6, TNF and nitrites in the supernatant of WT and Peli1-KO microglia cultures that left untreated (NT) or stimulated with LPS (100 ng/ml) at the indicated time points. (c,d) QPCR analysis of relative mRNA expression for the indicated inflammatory genes in the primary cultured microglia (c) or astrocytes (d) isolated from WT and Peli1-KO mice, left untreated (NT) or stimulated with IL-1β (20 ng/ml). The gene expression were normalized to a reference gene Actb (encoding β-actin) and showed as the relative value to Actb in bar graphs. Data with error bars represent mean ± SD. Each panel is representative of at least three independent experiment. *P < 0.05, **P < 0.01 as determined by two-way ANOVA with a Bonferroni post test.
Peli1 does not affect the inflammatory responses in astrocytes
Published studies showed that astrocytes have also been well-characterized to function as a key modulator of neuroinflammation during neurodegeneration26,27. In this model, astrocyte can be activated by the IL-1β and/or TNF that produced by activated microglia, and together with microglia, synergistically promote the neurotoxic effect25. Although Peli1 is known as a positive regulator of IL-1R signaling in innate immune cells16,17,18, we found that IL-1β-induced inflammatory responses are not affected in Peli1-deficient microglia and astrocyte as compared to WT cells (Fig. 4c,d). In addition, we have previously demonstrated Peli1 is dispensable for the inflammatory responses induced by TNF in astrocytes19. Together, these results demonstrated that the suppression of LPS-induced neuroinflammation in SN of Peli1-deficient mice is due to the defective activation of microglia, but not astrocyte.
Peli1 deficiency inhibits microglia-mediated neuron death
To confirm that Peli1-mediated microglial activation is responsible for the neuron death during LPS-induced SN inflammation, we examined the neurotoxicity to Neuro2A cells by using different conditioned medium (CM) that isolated from LPS-stimulated primary cultured WT or Peli1-deficient microglia or astrocyte (Fig. 5a). We also used TNF plus cycloheximide (CHX) stimulation as a positive control, which can induce Neuro2A cell death. As expected, WT microglia-derived CM significantly promoted the apoptosis of Neuro2A cells, whereas CM from Peli1-deficient microglia dramatically inhibited Neuro2A cell death. However, CM of astrocytes from both WT and Peli1-deficient mice didn’t affect the survival of Neuro2A cells, and there is no significant difference of these two groups. Interestingly, WT microglia-derived CM significantly promoted the production of neurotoxic factors of both WT and Peli1-deficient astrocytes. This effect was markedly inhibited when using the CM from Peli1-deficient microglia (Fig. 5b). More interestingly, CM from WT primary microglia also induced significant cell death of primary neurons, but Peli1 deficiency in microglia almost abolished the toxic effect on primary neurons that induced by microglia-derived inflammatory mediators (Fig. 5c).

Peli1 deficiency inhibited microglia-mediated neuron death. (a) Scheme of conditioned media (CM) and cell death assay by using WT and/or Peli1-KO microglia or astrocytes. (b,c) CMs were harvested from WT and Peli1-KO microglia that were stimulated with LPS (100 ng/ml) for 24 h, and then used for the test of viability of Neuro2A cells (b) or primary neurons (c). For sequential CM assay, CMs harvested from microglia were cultured with WT and Peli1-KO astrocytes for 24 h. Then, CMs were harvested and tested for effect on viability of Neuro2A cells. Neuro2A cells that treated with TNF (20 ng/ml) plus cycloheximide (CHX) were used as a positive control. (d) Effect of Peli1 knockdown in Neuro2A cells on sensitivity to TNF (20 ng/ml) plus CHX induced apoptosis assessed by flow cytometry assay, showing the frequencies of Annexin V+ cells in bar graph. Data with error bars represent mean ± SD. Each panel is representative of at least three independent experiment. *P < 0.05, **P < 0.01 as determined by one-way ANOVA with a Tukey’s post test (b–d).
To exclude the possibility that Peli1 in neurons may directly regulate the inflammation-induced cell death, we knocked down Peli1 gene expression in Neuro2A cells and stimulated with TNF plus CHX, a positive inducer of cell death. The results showed in Fig. 5d indicated that Peli1 knockdown in Neuro2A cells does not affect neuron cell death induced by TNF plus CHX. These results further confirmed that Peli1-mediated activation and production of neurotoxic mediators in microglia contribute to the neuron death during LPS-induced SN inflammation.
Peli1 mediates NF-κB and MAP kinase activation in microglia
To dissect the molecular mechanisms of Peli1-mediated microglial activation, we reduced Peli1 expression in murine BV2 microglia cells (Fig. 6a), and Peli1 knockdown does not affect the growth and survival of BV2 cells (Fig. 6b). We next investigated the function of Peli1 in modulating LPS-induced gene expression in Peli1-knockdown BV2 cells. The results revealed that the expression of Nos2 and proinflammatory cytokine genes were significantly inhibited in Peli1-knockdown BV2 cells in response to LPS stimulation (Fig. 6c), which confirmed the phenotype we discovered in primary microglia. Since NF-κB and MAP kniase activation play important role in LPS-mediated inflammation, we next tested the activation status of these signal pathways in BV2 cells. The results indicated Peli1 knockdown in BV2 cells significantly inhibited NF-κB activation by EMSA analysis, and MAP kinase activation by immunoblot analysis (Fig. 6d–f), which were consistent with our previously published data obtained from primary microglia. Taken together, Peli1 mediated LPS-induced activation of NF-κB and MAP kniase, and thus elevated the activation neurotoxic gene expression in microglia.

Peli1 mediated NF-κB and MAP kinase activation in microglia. (a) Immunoblot analysis of Peli1 showing the knockdown efficiency in murine BV2 microglial cells, Hsp60 immunoblot was used as a loading control. (b) MTT analysis examining the survival of control and Peli1-knockdown BV2 cells. (c) QPCR analysis of relative mRNA expression for the indicated inflammatory genes in the BV2 cells that infected with lentivirus encoding control (shCtrl) or Peli1 shRNA (shPeli1), left untreated (NT) or stimulated with LPS (100 ng/ml). The genes’ expression were normalized to a reference gene Actb (encoding β-actin) and showed as the relative value to Actb in bar graphs. (d) Electrophoretic mobility-shift assay (EMSA) of NF-κB in nuclear extracts of control or Peli1-knockdown BV2 cells left unstimulated or stimulated with LPS (100 ng/ml) at the indicated time points. Lamin B immunoblot was used as a loading control. (e,f) Immunoblot analysis of phosphorylated (P-) MAP kinases in whole-cell lysates of control or Peli1-knockdown BV2 cells (e) or in primary microglia (f) that left unstimulated or stimulated with LPS (100 ng/ml) at the indicated time points. The relative protein levels were quantified by ImageJ and the values were presented below each blot. Data with error bars represent mean ± SD. Each panel is representative of at least three independent experiment. **P < 0.01 as determined by unpaired Student’s t test (b) or two-way ANOVA with a Bonferroni post test (c).
Discussion
The specific loss of dopaminergic neurons in the SN is the hallmark of PD10,28. Until now, the detailed mechanism controlling dopaminergic neuronal cell death during PD pathogenesis are not fully understood. However, published studies have suggested that the local inflammation in SN may be function as an active contributor to promote the pathology of both human and mouse PD29,30,31. Therefore, an improved understanding of the mediators that regulate CNS inflammatory responses during PD pathogenesis will promote the understanding of the pathogenic mechanism of PD and thus help develop new therapeutic strategies. Here, we found that the E3 ubiquitin ligase Peli1 function as an important mediator to regulates the pathogenesis of PD. Loss of Peli1 significantly protected neurons from inflammation-induced neurotoxicity.
Microglial activation has been recognized as a critical event that initiates inflammatory responses in the pathogenesis of PD14,25. In the present study, we found that deficiency of Peli1 in primary cultured microglia dramatically suppressed LPS-induced secretion of neurotoxic mediators, leading to promoted neuron cell survival. However, Peli1 deficiency does not affect astrocyte activation by IL-1β stimulation, which is consistent with previous report that Peli1 is dispensable for the IL-1R signaling pathway in mouse embryo fibroblasts (MEF)32. In addition, LPS-mediated microglial activation and production of neurotoxic factor are sufficient to induce neuron death. Peli1 deficiency in microglia obviously inhibited this inflammation-induced cell death. More interestingly, LPS-induced pro-inflammatory cytokines by microglia further exhibit a paracrine function to activate astrocytes, which increase the production of neurotoxic factors, leading to exaggerated neurotoxic effect. Consistent with the gene expression results, Peli1 deficiency in astrocyte does not affect neuron death by astrocyte conditioned medium that stimulated with the same microglial conditioned medium. Collectively, our data further confirmed that microglial activation is a critical initiator of inflammation in PD pathogenesis.
The microglia-specific function of Peli1 may be attribute to its expression pattern in different glial cells. Our previous data have shown that astrocyte express three Peli family members (Peli1/2/3) at comparable levels19. Given the extremely high levels of sequence identity of the different Peli proteins, it is very likely that they have functional redundancies in IL-1R signaling functions in astrocyte. Due to the predominant expression of Peli1 in microglia, knockdown of Peli1 in BV2 cells not only affected the TRIF-mediated TLR signaling in NF-κB pathway, but also impaired the MyD88-dependent TLR signaling in the activation of MAPKs. These results are consistent with our previously findings in Peli1-deficient innate immune cells32 and primary microglia19.
Although no evidences indicated that LPS or bacterial infection directly involve in the human PD pathogenesis, very little LPS has been found in the brains under normal physiological condition33, suggesting a possibility that the entered LPS may initiate the neuroinflammation. Accumulating evidences suggested that aging increase the generation of endogenous TLR ligands, which may promote the disease initiation or progression34,35. In fact, repeated intraperitoneal injection of LPS exacerbates motor axon degeneration in the mouse system36,37, suggesting LPS or bacteria infection may be a risk factor to promote the neuroinflammation and thus induce the death of dopamine neurons. The present findings further confirmed that modulation of LPS-induced TLR inflammatory signaling by Peli1 in microglia indeed controlled the pathogenesis of PD.
In conclusion, we provided the evidences that Peli1 is induced in the SN of human and mouse PD brain, in which Peli1 facilitates microglial over-activation through NF-κB and MAP kinases. The activated microglia that produced amounts of neurotoxic factors, together with the inflammatory mediators produced by activated astrocytes, contributed to inflammation-induced death of dopaminergic neurons. Therefore, targeting Peli1 may suppress expression of neurotoxins, and thus have therapeutic effect in neuroinflammation and related neurological diseases.
Methods
Mice
Peli1-deficient mice (on the C57BL/6 background) were obtained as described previously21. Briefly, Peli1+/− heterozygous mice were bred to generate age-matched Peli1−/− (Peli1-KO) and Peli1+/+ (WT) mice. Mice were maintained in a specific pathogen-free facility, and all animal experiments were in accordance with protocols approved by the Institutional Animal Care and Use Committee of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences.
Antibodies and reagents
Antibodies for phospho-JNK (4668) and phospho-p38 (9215) were purchased from Cell Signaling Technology Inc. The anti-phospho-ERK1/2 (sc-7383), Peli1 (sc-271065), Hsp60 (sc-13115), iNOS (sc-7271) and Lamin B (sc-6216) antibodies were from Santa Cruz. The anti-TH antibody (MAB318) was from Chemicon, anti-Iba1 antibody (019-19741) was from WAKO, and anti-GFAP antibody (G3893) were from Sigma-Aldrich. LPS (L3129) was from Sigma-Aldrich. The cycloheximide (CHX) (HY12320) was from MCE. Murine IL-1β (211-11B) and TNF (315-01 A) were from Peprotech.
PELI1 gene expression analysis of human samples
To analyze the relative PELI1 expression in human samples, we extracted the data from NCBI GEO database (GDS2821, GDS3128, GDS3129, GDS4145, GDS2519), and compared the expression of PELI1 in the PBMCs or SN from health donors and PD patients.
LPS-induced PD mouse model
The induction of PD mouse model was as described previously25. Briefly, WT and Peli1-defiicient 8-10-week-old male mice (n = 4–5 mice/group) were anesthetized and then immobilized in a stereotaxic apparatus. Two microliters of LPS (5 μg/ml) or equal volume of PBS were stereotaxically injected with into the SN (AP, −3.3 mm; ML, ±1.2 mm; and DV, −4.6 mm) over a 5-minute period by using a stainless-steel syringe (Hamilton). Mice were euthanized 6 h after the stereotaxic injection, and the injected SN was dissected under a dissection microscope and the tissue was processed for quantitative PCR. Seven days after the injection, the mice were sacrificed, and the brain slices were collected for immunohistochemistry analysis.
Immunohistochemistry (IHC) and immunofluorescence (IF)
The IHC or IF staining was as described previously25,26. Experimental mice were anesthetized and perfused transcardially with PBS followed by 4% paraformaldehyde. Brain samples were postfixed with 4% paraformaldehyde overnight and equilibrated in 30% sucrose. Coronal sections of 10 μm were prepared with a sliding microtome, and were then incubated with primary antibodies: rabbit anti-tyrosine hydroxylase pAb (1:500; Chemicon); rabbit anti-Iba1 pAb (1:500; WAKO); rabbit anti-GFAP pAb (1:800; DAKO); mouse anti-GFAP mAb (1:1,000, Sigma-Aldrich). The brain slices were then incubated with the horseradish peroxidase (HRP)- or fluorescence-conjugated secondary antibodies. The peroxidase activity of immune complexes was revealed with a DAB kit according to the manufacturer’s instruction (Beyotime, P0203). Sections were imaged using either a cooled CCD (DP72, Olympus) on a microscope (BX51; Olympus).
Cell quantification
The number of tyrosine-hydroxylase (TH)-positive cells was quantified in adult Peli1-KO and their littermates in brain cryosections with typical morphology of the substantia nigra as previously described26. In brief, four series of cryosections were collected and every fourth section (10 μm) was used for quantification of TH-positive neurons. The average intensities of GFAP or Iba1 were calculated using ImageJ in the substantia nigra.
Primary glial cell culture
Mixed glial cultures were prepared from neonatal mice that age around 1–2 days as described previously26. In brief, after removing the meninges, the neonatal brains were dissociated by 0.25% trypsin, filtered with a 40-μm mesh, and the dissociated cells were plated in 10 cm dish in DMEM/ F12 medium containing 10% FBS, penicillin and streptomycin at 37 °C in humidified 5% CO2/95% air. Culture media were changed twice a week. The mixed glial cells were reached to 90% confluence at around day 9, were then re-plated after trypsinization. At day 20 in vitro, cultures were mildly trypsinized with trypsin solution (0.07% trypsin in DMEM/Ham’s F12) at 37 °C for 15–20 min. Floating cells (astrocytes and dead cells) were removed by rinsing cultures with D-Hanks’ solution. The resulting enriched microglial cultures were maintained in DMEM/Ham’s F12 complete medium containing 10% FBS, penicillin and streptomycin until use. The purity of the microglia was >97% as determined by flow cytometry to measure the percentage of CD11b+ cells.
Primary neuron culture
The primary mouse neurons were prepared by following our previous protocol19. Briefly, we dissociated the cortex of newborn mouse brains in 0.25% trypsin, filtered the cell suspension with a 40 μm mesh and cultured the cells in Neurobasal medium (Invitrogen) supplemented with B-27 (Invitrogen), 100 U/ml penicillin and 100 mg/ml streptomycin. The culture medium was changed every 3 d for 2 weeks. The collected primary neurons were then applied for the survival assay by using the CM from LPS-stimulated primary microglia.
Quantitative RT-PCR
Brain tissues or cell samples were homogenized in Trizol reagent26. cDNA was synthesized from 1 μg of extracted total RNA using M-MLV Reverse Transcriptase kit (Takara) according to the manufacturer’s instructions. Quantitative PCR was performed with SYBR-Green premix ExTaq (Roche) and detected by a Real Time PCR System by using gene-specific primer sets (Table 1). The relative genes’ expression was assessed in triplicate, normalized to a reference gene Actb (encoding β-actin) and determined based on 2−∆∆Ct method38.
Measurement of the inflammatory cytokines and nitrite
The concentrations of mouse IL-1β, TNF and IL-6 in the supernatants of primary microglial cultures that with or without stimulation were measured through enzyme-linked immunosorbent assay (ELISA). Briefly, the 96-well flat plates were coated with the capture antibodies for IL-1β (eBioscience, 14-7012-81), TNF (eBioscience, 14-7325-85) or IL-6 (eBioscience, 14-7061-85) at 4 °C overnight. The microplates were blocked with 3% BSA, incubated with the supernatants at room temperature for 2 h, and followed by incubation with biotinylated detection antibodies for IL-1β (eBioscience, 13-7112-81), TNF (eBioscience, 13-7341-85) or IL-6 (eBioscience, 13-7062-85). Then HRP-conjugated avidin (eBioscience, 18-4100-51) were added into each well for 30 min at room temperature, visualized by TMB solution (eBioscience, 00-4201-56) and quantified through a TECAN ELISA reader. The nitrite concentration in the supernatants were examined by using a NO assay kit (Beyotime, S0021) according to the manufacturer’s instruction.
Immunoblot (IB) and electrophoresis mobility shift assays (EMSA)
IB analysis were performed as previously described19. Primary microglia were stimulated with 100 ng/ml LPS for the indicated time period and lysed in RIPA buffer. The whole cell extracts were separated via 8.25% SDS-PAGE, transferred to PVDF membranes, blocked and subject to IB analysis. Nuclear extract preparation and EMSAs were carried out as described previously19.
Statistical analysis
Statistical analysis was performed as previously described39. Differences between groups were established using a Student’s unpaired t test (for two groups), a one-way ANOVA with a Tukey’s post test for multiple groups’ comparisons, or a two-way ANOVA with a Bonferroni post test when comparing groups with multiple variables. Data are presented as means ± SD. A P-value less than 0.05 is considered statistically significant. Statistic details are indicated in the respective figure legends.
Ethics approval and consent to participate
All study surgical procedures and experiment protocols were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China).
References
Farrer, M. J. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet 7, 306–318 (2006).
Fearnley, J. M. & Lees, A. J. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114, 2283–2301 (1991).
Block, M. L., Zecca, L. & Hong, J. S. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8, 57–69 (2007).
Nguyen, M. D., Julien, J. P. & Rivest, S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci 3, 216–227 (2002).
Liu, B. et al. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann N Y Acad Sci 962, 318–331 (2002).
Brown, G. C. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem Soc Trans 35(Pt 5), 1119–1121 (2007).
Brown, G. C. & Neher, J. J. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol 41, 242–247 (2010).
Hartmann, A., Hunot, S. & Hirsch, E. C. Inflammation and dopaminergic neuronal loss in Parkinson’s disease: a complex matter. Exp Neurol 184, 561–564 (2003).
Teismann, P. & Schulz, J. B. Cellular pathology of Parkinson’s disease: astrocytes, microglia and inflammation. Cell Tissue Res 318, 49–61 (2004).
Arai, H., Furuya, T., Mizuno, Y. & Mochizuki, H. Inflammation and infection in Parkinson’s disease. Histol Histopathol 21, 673–678 (2006).
Gao, H. M. et al. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson’s disease. J Neurochem 81, 1285–1297 (2002).
Gibbons, H. M. & Dragunow, M. Microglia induce neural cell death via a proximity-dependent mechanism involving nitric oxide. Brain Res 1084, 1–15 (2006).
Meredith, G. E., Sonsalla, P. K. & Chesselet, M. F. Animal models of Parkinson’s disease progression. Acta Neuropathol 115, 385–98 (2008).
Saijo, K., Collier, J. G., Li, A. C., Katzenellenbogen, J. A. & Glass, C. K. An ADIOL-ERβ-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell 145, 584–595 (2011).
Kawai, T. & Akira, S. TLR signaling. Semin Immunol 19, 24–32 (2007).
Jin, W., Chang, M. & Sun, S. C. Peli: a family of signal-responsive E3 ubiquitin ligases mediating TLR signaling and T-cell tolerance. Cell Mol Immunol 9, 113–122 (2012).
Moynagh, P. N. The Pellino family: IRAK E3 ligases with emerging roles in innate immune signalling. Trends Immunol 30, 33–42 (2009).
Schauvliege, R., Janssens, S. & Beyaert, R. Pellino proteins: novel players in TLR and IL-1R signalling. J Cell Mol Med 11, 453–461 (2007).
Xiao, Y. et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat Med 19, 595–602 (2013).
Chang, M. et al. The ubiquitin ligase Peli1 negatively regulates T cell activation and prevents autoimmunity. Nat Immunol 12, 1002–1009 (2011).
Liu, J. et al. Peli1 negatively regulates noncanonical NF-κB signaling to restrain systemic lupus erythematosus. Nat Commun 9, 1136 (2018).
Xiao, Y. et al. Peli1 negatively regulates type I interferon induction and antiviral immunity in the CNS. Cell Biosci 5, 34 (2015).
Brochard, V. et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119, 182–192 (2009).
Sampson, T. R. et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 167, 1469–1480 (2016).
Saijo, K. et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137, 47–59 (2009).
Shao, W. et al. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via αB-crystallin. Nature 494, 90–94 (2013).
Xiao, Y. et al. TPL2 mediates autoimmune inflammation through activation of the TAK1 axis of IL-17 signaling. J Exp Med 211, 1689–1702 (2014).
Chung, Y. C. et al. The role of neuroinflammation on the pathogenesis of Parkinson’s disease. BMB Rep 43, 225–232 (2010).
McGeer, P. L., Itagaki, S., Boyes, B. E. & McGeer, E. G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291 (1988).
Mogi, M. et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett 180, 147–150 (1994).
Mogi, M. et al. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci Lett 211, 13–16 (1996).
Chang, M., Jin, W. & Sun, S. C. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat Immunol 10, 1089–1095 (2009).
Nadeau, S. & Rivest, S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience 93, 1449–1464 (1999).
Di Virgilio, F. Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol Sci 28, 465–472 (2007).
Lehnardt, S. et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA 100, 8514–8519 (2003).
Ling, Z. et al. In utero bacterial endotoxin exposure causes loss of tyrosine hydroxylase neurons in the postnatal rat midbrain. Mov Disord 17, 116–124 (2002).
Nguyen, M. D., Julien, J. P. & Rivest, S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci 3, 216–227 (2002).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Lucin, K. M. et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 79, 873–786 (2013).
Acknowledgements
We gratefully acknowledge the technical support from Dr. Jiawei Zhou’ lab for the construction of PD mouse models, and thank the animal facility for the animal care and maintenance. This research was supported by the grants from the Jiangsu Provincial Key and Development Program (BE2016722), the National Natural Science Foundation of China (81571545, 81770567), the Thousand Young Talents Plan of China, CAS Key Laboratory of Tissue Microenvironment and Tumor.
Author information
Authors and Affiliations
Contributions
D.D., J.X., J.Y. and Y.W. performed the experiments, prepared the figures, and wrote parts of the manuscript; C.M. contributed to the experiments and data analysis; Y.X. designed and supervised the research, and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dai, D., Yuan, J., Wang, Y. et al. Peli1 controls the survival of dopaminergic neurons through modulating microglia-mediated neuroinflammation. Sci Rep 9, 8034 (2019). https://doi.org/10.1038/s41598-019-44573-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44573-w
This article is cited by
-
The Role of Ubiquitin–Proteasome System and Mitophagy in the Pathogenesis of Parkinson's Disease
NeuroMolecular Medicine (2023)
-
Neuroprotective action of α-Klotho against LPS-activated glia conditioned medium in primary neuronal culture
Scientific Reports (2022)
-
The ubiquitin ligase Peli1 inhibits ICOS and thereby Tfh-mediated immunity
Cellular & Molecular Immunology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
