
Abstract
Several placebo-controlled trials have been recently published evaluating novel therapies targeting the defective CFTR protein. This systematic review examines the clinical efficacy and safety of CFTR modulators in individuals with cystic fibrosis (CF) with specific genetic mutations. Online sources were searched for placebo-controlled, parallel-design clinical trials investigating CFTR modulators from January 1, 2005 to March 31, 2018. The primary outcome of interest was FEV1% predicted (ppFEV1). Fourteen RCTs met our eligibility criteria. The largest improvement in ppFEV1 favouring treatment was observed for ivacaftor (IVA) in G551D individuals (≥6 years old). Both tezacaftor-ivacaftor (TEZ-IVA) and lumacaftor-ivacaftor (LUM-IVA) also improved ppFEV1 in F508del homozygous individuals but there was increased reporting of respiratory adverse events with LUM-IVA compared to placebo. IVA also significantly improved ppFEV1 in a sub-group of individuals ≥18 years old with an R117H mutation. No significant improvements in ppFEV1 were observed for IVA, LUM, or TEZ in F508del homozygous individuals, LUM or LUM-IVA in F508del heterozygous individuals, or ataluren in individuals with a nonsense mutation. Significant improvements in ppFEV1 and other clinical outcomes were observed for IVA in G551D individuals, TEV-IVA and LUM-IVA in F508del homozygous individuals, and IVA in adults with a R117H mutation.
Similar content being viewed by others

Introduction
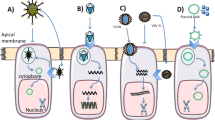
Cystic fibrosis (CF) is a genetic condition caused by dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) protein. CFTR is located at the apical surface of epithelial cells and the absence of CFTR activity leads to loss of chloride secretion and deficient fluid transport1. This results in thick and sticky secretions involving a range of epithelial tissues such as the airways and pancreatic ducts, eventually culminating in end-organ damage and failure. Since the discovery of the CFTR gene in 19892, significant progress has been made in the understanding of how CFTR gene mutations alter protein structure and function leading to reduced CFTR activity3.
Although over 2000 variants in the CFTR gene have been identified to date, F508del accounts for most CFTR alleles in patients with CF. This particular mutation leads to abnormal CFTR folding and trafficking causing reduced delivery of CFTR to the cell surface4. Another class of CFTR mutations, referred to as “nonsense” mutations, leads to a premature termination codon and reduced synthesis and hence delivery of CFTR to the cell surface5. In contrast, “gating” mutations are missense mutations that lead to CFTR proteins that are sufficiently synthesized, processed and trafficked to the cell surface but once they arrive they have defective channel opening leading to diminished chloride secretion6.
With advances in our understanding of CFTR biology, a new class of small molecule therapies, referred to as CFTR modulators, have been identified using high-throughput small molecule screening; these drugs are unique as they directly target molecular defects in the CFTR protein to increase CFTR activity7,8,9,10,11. For example, CFTR “potentiators” are small molecules capable of increasing the amount of time the CFTR channel is spent in the open position and thus targets CFTR mutations with defective “gating”10. CFTR “correctors” are small molecules that can target mutations such as F508del as they can improve CFTR trafficking or transport to the cell surface by stabilizing the 3D conformation of the protein, even if misfolded11. Other CFTR modulators, including CFTR “amplifiers” and “translational read-through” agents increase the amount of CFTR protein produced, the latter being specific to mutations leading to a premature termination codon12,13.
In recent years, several placebo-controlled clinical trials have been conducted investigating the efficacy and safety of CFTR modulators but the results have varied depending on the specific CF genotype and therapy under investigation8. The primary objective of this systematic review was to evaluate the impact of CFTR modulators on lung function and other clinically important outcomes including pulmonary exacerbations, hospitalizations, respiratory symptoms, nutritional status, and adverse events in individuals with CF.
Methods
Search strategy
Our search strategy was developed in accordance with PRISMA guidelines14. A systematic search of online databases using key phrases was conducted to identify randomized, placebo-controlled trials published from January 1, 2005 to March 31, 2018. Online databases searched included: MEDLINE, EMBASE, ACP Journal Club, Cochrane Central Register for Controlled Trials (CENTRAL), Cochrane Database of Systematic Reviews (CDSR), Cochrane Methodology Register (CMR), Database of Abstracts of Reviews of Effects (DARE), Health Technology Assessment (HTA), and NHS Economic Evaluation Database (NHSEED). For comprehensiveness, clinical trial registries such as the European Medicines Agency, U.S. National Institute of Health, and the World Health Organization records were accessed and screened. We used the following key phrases which were designed to maximize sensitivity for detecting therapeutic trials in CF: (“cystic fibrosis” OR “CFTR”) AND (“drug therapy” OR “clinical trial”).
Selection criteria
The literature search and abstracts were reviewed for eligibility independently by two investigators (A.R.H and M.K.). Randomized controlled trials (RCTs) with a parallel design comparing CFTR modulators (e.g. potentiators, correctors, translational read-through agents) to placebo in patients with CF were included. Study inclusion/exclusion were summarized in a PRISMA flow diagram14. The level of agreement in the articles selected for full text review and then for inclusion in the review by the two investigators were reported and discrepancies were resolved by the principal investigator (B.S.Q).
Data extraction
The review protocol used in this study is available in the Appendix and was developed in accordance with the PRISMA statement14. Two reviewers (A.R.H. and M.K.) independently extracted data. The level of agreement in the data extracted broken down by study characteristics, risk of bias, and effects of the intervention by the two investigators were reported and discrepancies were resolved by the principal investigator (B.S.Q).
Risk of bias assessment
Risk of bias was assessed using the Cochrane Risk of Bias tool15. A detailed review of the randomization process, blinding, and allocation sequence concealment was performed.
Outcomes
Change in percent-predicted forced expiratory volume in one second (ppFEV1) was our primary outcome. Secondary efficacy outcomes included protocol-defined pulmonary exacerbations (PEx), hospitalization due to PEx, respiratory symptoms (i.e., Cystic Fibrosis Questionnaire-Revised (CFQ-R) Respiratory domain), and nutritional status (i.e., body mass index and weight). Adverse events with a prevalence of >10% (and involving >2 subjects) from either experimental or control groups, serious adverse events (including deaths) leading to treatment discontinuation, and the prevalence of elevated liver function tests (LFTs) were evaluated.
Statistical analysis
The statistical analysis was performed using ReviewManager (RevMan 5.3, Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014) in accordance with the Cochrane Handbook16. For each clinical outcome, the results were stratified by genotype and type/dose of CFTR modulator. If two or more studies evaluated the same drug at the same dose in the same genotype, the data was pooled using a fixed-effect meta-analysis (Appendix). For the primary outcome, ppFEV1, sub-group analyses were planned based on age and baseline ppFEV1.
Results
Study selection
The search yielded a total of 789 potentially relevant articles and abstracts. Following full-text review, thirteen articles (14 placebo-controlled, parallel-group studies) met the inclusion and exclusion criteria (Fig. 1).

Characteristics of included studies
A total of eight phase 3 and six phase 2 studies from thirteen original articles were identified. The article by Wainwright et al. included two phase 3 studies accounting for the discordance between the number of articles and studies17. The proposed class/mechanism of action for each CFTR modulator along with the number of studies evaluating the therapy is described in Table 1. Characteristics of the included studies and its participants are detailed in Table 2 and Appendix Table 1. The a priori outcomes of interest for the included studies are summarized in Appendix Table 2.
Risk of bias of included studies
Risk of bias for each included article is summarized in Appendix Fig. 1. Most studies were considered ‘low risk’ for selection, performance, and attrition bias (Fig. 2)17,18,19,20.

Risk of Bias Summary for Included Studies. Selective outcome reporting was noted for Kerem et al.18 as the study authors did not report in their full text publication all outcomes listed in their study protocol including antibiotic use and hospitalization due to CF-related symptoms, disruption to school or work due to CF-related symptoms, and pharmacokinetics. Similarly, Ramsey et al.20 did not report on all CFQ-R domain items or tertiary outcomes pre-defined in their clinical trial protocol including EQ-5D, oxygen saturation, and outpatient sick visits to the clinic or hospital for CF-related complications. Ratjen et al.19 did not report data on exacerbations (time to first, number) and the Treatment Satisfaction Questionnaire despite these being listed as secondary endpoints in the publication. Wainwright et al.17 did not report data on the EQ-5D or Treatment Satisfaction Questionnaire despite it being listed in their trial protocol.
Effects of the intervention
Primary outcome
ppFEV1: Of all the CFTR modulators examined to date, individuals with a G551D mutation treated with IVA experienced the largest improvement in ppFEV1 compared to placebo (n = 2 studies; n = 213; weighted absolute mean difference 10.8, 95% CI: 9.0–12.7) (Fig. 3A) with no heterogeneity (I2 = 0%) in results between studies (Fig. 3B)20,21.

Absolute Difference in ppFEV1 for Patients Randomized to CFTR Modulators vs. Placebo. (A) Data from individual studies; (B) Meta-analysis combining data if identical CFTR modulator and dose. Footnote: (1) Individuals received IVA at baseline as part of routine clinical care and therefore the control group received IVA + Placebo. Abbreviations: D1–14 = day 1 to day 14; D1–21 = day 1 to day 21; D1–28 = day 1 to day 28; D1–56 = day 1 to day 56; IVA = ivacaftor; LUM = lumacaftor; TEZ = tezacaftor; ^2 = twice a day.
For F508del homozygous individuals 12 years and older, ppFEV1 significantly improved with LUM-IVA and TEZ-IVA compared to placebo (Fig. 3A). The effect size was similar for TEZ-IVA (n = 2 studies; n = 535; weighted absolute mean difference 4.0, 95% CI: 3.2–4.8)22,23 and higher dose LUM-IVA (n = 3 studies; n = 755; weighted absolute mean difference 3.4, 95% CI: 2.4–4.4) (Fig. 3B)17,24. For individuals 6–11 years, there was a mild increase in ppFEV1 for LUM-IVA compared to placebo (n = 1 study; n = 204; absolute mean difference 2.4, 95% CI: 0.4–4.4)19. No significant treatment effect was observed with IVA or TEZ alone, and there was a trend toward worsening in ppFEV1 for F508del homozygous individuals treated with higher doses of LUM (Fig. 3A)22,24,25.
For F508del heterozygous individuals, there was no significant improvement in ppFEV1 on LUM or LUM-IVA (Fig. 3A)24,26. In a small study involving individuals with F508del/G551D, TEZ-IVA did not lead to a significant improvement in ppFEV1 compared to IVA alone22.
For individuals with the R117H mutation on at least one allele, IVA did not lead to an overall improvement in ppFEV1 compared to placebo, but there was a significant improvement in a pre-defined subgroup analysis restricted to adults (n = 50; absolute mean difference 5.0, 95% CI 1.2–8.8)27. For individuals with a nonsense mutation on at least one allele, ataluren did not result in a significant relative improvement in ppFEV1 compared to placebo18.
Secondary outcomes
Pulmonary exacerbations (PEx): Eight studies examined protocol-defined PEx as described in Appendix Table 3. Of all the CFTR modulators examined, individuals (≥12 years old) with a G551D mutation receiving IVA derived the greatest reduction in PEx risk compared to placebo (n = 1 study; n = 161; OR 0.39, 95% CI: 0.21–0.74) (Appendix Fig. 2A)20. LUM-IVA and TEZ-IVA also significantly reduced the risk of PEx compared to placebo in F508del homozygous individuals (≥12 years old) but the risk reduction was less than that observed with IVA in G551D (Appendix Fig. 2A,B)17,23. In comparison to placebo, no significant reduction in PEx risk was observed for F508del homozygous individuals or individuals with the R117H mutation on at least one allele receiving IVA, nor for individuals with a nonsense mutation receiving ataluren (Appendix Fig. 2A)18,25,27.
Pulmonary exacerbations (PEx) requiring hospitalization: LUM-IVA reduced the risk of PEx requiring hospitalization in F508del homozygous individuals (Appendix Fig. 3A,B)17. TEZ-IVA also significantly reduced the rate of PEx leading to hospitalization compared to placebo (n = 1 study; n = 504; rate ratio 0.53, 95% CI 0.34–0.82) but a risk ratio could not be calculated23. Individuals with the G551D mutation on at least one allele treated with IVA also experienced a reduction in the risk of PEx requiring hospitalization but this was not statistically significant (Appendix Fig. 3A)20.
CFQ-R respiratory domain: Compared to placebo, CFQ-R Respiratory domain scores improved to a similar extent for IVA treated individuals (≥6 years old) with the G551D mutation on at least one allele (n = 3 studies; n = 236; weighted absolute mean difference: 7.2, 95% CI: 3.3–11.1)20,21,28, IVA treated individuals ≥18 years old with at least one R117H mutation (n = 1 study; n = 69; absolute mean difference: 8.4, 95% CI: 2.2–14.6)27, and for LUM-IVA treated F508del heterozygous individuals ≥18 years old (n = 1 study; n = 125; absolute mean difference: 6.5, 95% CI 1.4–11.6) (Appendix Fig. 4A,B). CFQ-R Respiratory domain scores also significantly improved with TEZ-IVA and LUM-IVA in F508del homozygous individuals (≥12 years old) but the mean difference did not exceed the minimal clinically important difference (MCID) for LUM-IVA17,23,24. Furthermore, there was no significant improvement in CFQ-R Respiratory domain scores for patients 6–11 years old on LUM-IVA compared to placebo19.
There was worsening of the CFQ-R Respiratory domain score for F508del homozygous and heterozygous individuals (≥18 years old) on LUM alone (Appendix Fig. 4A)24. In a small phase 2 study involving individuals with F508del/G551D, TEZ-IVA did not lead to significant improvement in the CFQ-R Respiratory domain compared to IVA alone22. For individuals with a nonsense mutation on at least one allele, ataluren did not modify CFQ-R Respiratory domain score compared to placebo18.
Nutritional outcomes (BMI and weight): For individuals with at least one G551D mutation (≥6 years old), significant improvements in weight were observed on IVA compared to placebo (n = 2 studies; n = 213; weighted absolute mean difference: 2.8 kg, 95% CI: 1.8–3.8) (Appendix Fig. 5A,B)20,21. For F508del homozygous individuals (≥12 years old), a clinically modest but statistically significant increase in BMI was observed for both doses of LUM-IVA compared to placebo (Appendix Fig. 6A,B)17; however, no significant treatment effect was seen in individuals 6–11 years on LUM-IVA (Appendix Fig. 6A)19. TEZ-IVA did not lead to improvement in BMI compared to placebo in individuals 12 years and older (Appendix Fig. 6A)23. For F508del heterozygous individuals (≥18 years old), LUM-IVA did not result in significant improvement in weight or BMI compared to placebo26. There were no significant improvements in BMI compared to placebo among IVA treated individuals with an R117H mutation (Appendix Fig. 6A) or ataluren treated individuals with a nonsense mutation (data not shown)18,27.
Adverse event reporting: CFTR modulators were generally well tolerated compared to placebo (Appendix Figs 7–30). For studies involving F508del homozygous and heterozygous individuals, those assigned to LUM had increased dyspnea and “abnormal respiration” compared to placebo (Appendix Figs 11 and 13). F508del homozygous and heterozygous subjects assigned to LUM and LUM-IVA also had more respiratory-related adverse events leading treatment discontinuation compared to placebo (Appendix Table 4)17,24. For the one study involving individuals with a nonsense mutation, subjects receiving ataluren had increased incidence of acute kidney injury compared to placebo (15% vs. <1%) resulting in higher rates of treatment discontinuation18.
The prevalence of LFT abnormalities was generally similar between treatment and placebo, however there were a few exceptions. A greater proportion of G551D patients had severe ALT elevations (>8x ULN) on IVA compared to placebo (3.6% vs 0%) (Appendix Table 5)20. Milder elevations in AST (2–3X ULN) were observed for G551D patients on IVA and ALT or AST (>3X ULN) in F508del homozygous children aged 6–11 on LUM-IVA compared to placebo (Appendix Table 5)19,20.
Level of agreement for study selection and data extraction: There was a strong level of agreement (95%) for the articles selected between the two reviewers for full text review and 100% agreement between the two reviewers for the articles meeting eligibility criteria for inclusion in this review. The level of agreement for data extraction were as follows: study characteristics (n = 88 data points, 95% agreement), risk of bias (n = 92 data points, 84% agreement), and effects of the intervention (n = 480 data points, 81% agreement).
Discussion
This study represents the most comprehensive systematic review of the efficacy and safety of CFTR modulators performed to date. While evidence-based recommendations for the use of CFTR modulators were recently published and provides a valuable resource for practicing clinicians, this review provides a more concise and up-to-date synthesis of all the placebo-controlled clinical trial data29. No prior systematic review has compared all investigational CFTR modulators from phase 2 and 3 RCTs in specific CF genotypes30,31,32.
As this review highlights, patients with gating mutations such as G551D benefit the most from current CFTR modulators and those that are F508 homozygous have moderate benefit in comparison. Based on published parallel design trials, CFTR modulators have not been effective in F508 heterozygotes or those with nonsense mutations. However, in a recent phase 3 cross-over study evaluating IVA and TEZ-IVA in individuals ≥12 years old with F508del and a residual CFTR function mutation, improvements in ppFEV1 of 4.7% and 6.8%, respectively, were observed compared to placebo33. Furthermore, unpublished phase 2 data evaluating TEZ-IVA in combination with “next-generation” corrector molecules have demonstrated significant improvements in ppFEV1 in subjects with F508del and a minimal CFTR function mutation, some of whom have nonsense mutations.
When comparing the efficacy of CFTR modulators across all genotypes for ppFEV1, CF individuals (≥6 years old) with the G551D mutation on at least one allele receiving IVA experienced the largest benefit20,21. F508del homozygous subjects receiving TEZ-IVA (≥12 years old) and LUM-IVA (≥6 years old) also had improvements in ppFEV1 compared to placebo but the effect sizes were modest compared to IVA in G551D17,19,24. Individuals (≥18 years old) with the R117H mutation on at least one allele treated with IVA experienced similar improvement in ppFEV1 to F508del homozygous subjects treated with TEZ-IVA and LUM-IVA.
Similar to ppFEV1, the effect of CFTR modulators on PEx risk and respiratory symptoms were most pronounced with IVA in G551D adolescents and adults (≥12 years old), with a 60% reduction in PEx risk and a 7-point improvement in the CFQ-R Resp domain20,21. F508del homozygous adolescents and adults also had a 40–45% reduction in PEx risk on TEZ-IVA and LUM-IVA. While F508del homozygous subjects experienced improvements in the CFQ-R Resp domain on both TEZ-IVA and LUM-IVA, this was not clinically significant for LUM-IVA. Individuals with a R117H mutation also experienced improvements in the CFQ-R Resp domain on IVA, with a magnitude of change in the adults comparable to that observed with IVA in G551D. The effect of CFTR modulators on weight were most significant with IVA in G551D individuals (≥6 years old). While F508del homozygous individuals (≥12 years old) had improvement in BMI with LUM-IVA, the effect size was modest.
Most of the CFTR modulator therapies examined in this review were well tolerated with the exception of increased reporting of respiratory adverse events (e.g. dyspnea) leading to higher rates of treatment discontinuation in patients randomized to LUM and LUM-IVA. The molecular mechanism responsible for the adverse respiratory effects (e.g. dyspnea, abnormal respiration) for patients on LUM remain unclear but appears to be an off-target effect specific to LUM, as opposed to being related to F508del CFTR correction per se, as similar adverse effects have not been observed with F508del CFTR correction with TEZ-IVA22,23,34. There was also increased reporting of acute kidney injury for nonsense mutation patients assigned to ataluren compared to placebo. The long-term safety of CFTR modulator therapies beyond one year could not be assessed in this review and therefore the detection of infrequent or long-term side effects will require ongoing post-marketing surveillance35,36.
There are several potential limitations of this review. We excluded cross-over, open-label, and observational studies to avoid carryover effects and to ensure we incorporated the highest level of evidence. We also limited our inclusion to full-text studies which could have resulted in publication bias. We focused on pre-defined clinically important outcomes but did not include multiple-breath washout measurement (e.g. LCI2.5) given the lack of clinical trials utilizing this outcome measure19.
There remain several gaps in the placebo-controlled evidence base for CFTR modulators. RCTs to date have excluded young children (<6 years old) and therefore the earliest age of safe use of CFTR modulators remains uncertain. However, small open-label 24-week studies have demonstrated a similar safety profile of IVA in children 1–5 years old with CFTR gating mutations compared to older age groups studied37,38. Most RCTs have also excluded CF individuals with severe lung disease (ppFEV1 < 40%), individuals colonized/infected with bacteria associated with rapid lung function decline (e.g. Burkholderia cenocepacia, Mycobacterium abscessus), and individuals with very frequent pulmonary exacerbations requiring continuous or near continuous systemic antibiotics by virtue of requiring clinical stability and no systemic antibiotics 4 weeks prior to randomization and therefore the efficacy and safety of CFTR modulators in these sub-groups remain unclear. For example, based on observational data, F508del homozygous individuals with advanced lung disease started on LUM-IVA have increased respiratory-related adverse events leading to treatment discontinuation; therefore, closer monitoring following treatment initiation is recommended39,40.
Most placebo-controlled RCTs to date have been limited to a maximum duration of 48 weeks and therefore the long-term placebo-controlled effects of these therapies remain unclear. However, an open-label extension trial evaluating the long-term effects of ivacaftor up to 144 weeks has demonstrated sustained clinical benefits of ivacaftor on lung function, weight, patient-reported respiratory symptoms and PEx risk reduction with no new safety concerns35. Furthermore, based on combined data from an open-label extension trial and U.S. CF patient registry data, the rate of lung function decline over 3 years was lower in G551D patients treated with ivacaftor compared to propensity-matched controls from the CF registry, suggestive of a disease-modifying effect over the longer term.
In conclusion, based on randomized placebo-controlled parallel design trials, CFTR potentiation with IVA in individuals with a G551D mutation is safe, and results in robust clinical benefits compared to placebo and to date is superior to the effects observed with CFTR modulators in other CF genotypes. The effects of TEZ-IVA and LUM-IVA in F508del homozygous individuals are comparable with respect to the magnitude of change in ppFEV1 and PEx risk reduction but TEZ-IVA is safer and leads to greater improvement in respiratory symptoms.
References
Bear, C. E. et al. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 68(4), 809–18 (1992).
Riordan, J. R. et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245(4922), 1066–73 (1989).
Sheppard, D. N. & Welsh, M. J. Structure and function of the CFTR chloride channel. Physiol Rev 79(1 Suppl), S23–45 (1999).
Welch, W. J. Role of quality control pathways in human diseases involving protein misfolding. Semin Cell Dev Biol 15(1), 31–8 (2004).
Amrani, N. et al. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432(7013), 112–8 (2004).
Li, C. et al. ATPase activity of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 271(45), 28463–8 (1996).
Elborn, J. S. Personalised medicine for cystic fibrosis: treating the basic defect. Eur Respir Rev 22(127), 3–5 (2013).
Quon, B. S. & Rowe, S. M. New and emerging targeted therapies for cystic fibrosis. BMJ 352, i859 (2016).
Armstrong, D. K., Cunningham, S., Davies, J. C. & Alton, E. W. Gene therapy in cystic fibrosis. Arch Dis Child 99(5), 465–8 (2014).
Van Goor, F. et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 106(44), 18825–30 (2009).
Van Goor, F. et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA 108(46), 18843–8 (2011).
Molinski, S. V. et al. Orkambi(R) and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol Med 9(9), 1224–43 (2017).
Howard, M., Frizzell, R. A. & Bedwell, D. M. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med 2(4), 467–9 (1996).
Moher, D., Liberati, A., Tetzlaff, J., Altman, D. G. & Group, P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 6(7), e1000097 (2009).
Higgins, J. P. et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ 343, d5928 (2011).
Higgins J. P. & Green S. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, Available from, http://handbook.cochrane.org/ (2011).
Wainwright, C. E. et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med 373(3), 220–31 (2015).
Kerem, E. et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med 2(7), 539–47 (2014).
Ratjen, F. et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med 5(7), 557–67 (2017).
Ramsey, B. W. et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 365(18), 1663–72 (2011).
Davies, J. C. et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med 187(11), 1219–25 (2013).
Donaldson, S. H. et al. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med 197(2), 214–24 (2018).
Taylor-Cousar, J. L. et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med 377(21), 2013–23 (2017).
Boyle, M. P. et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med 2(7), 527–38 (2014).
Flume, P. A. et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 142(3), 718–24 (2012).
Rowe, S. M. et al. Lumacaftor/Ivacaftor Treatment of Patients with Cystic Fibrosis Heterozygous for F508del-CFTR. Ann Am Thorac Soc 14(2), 213–9 (2017).
Moss, R. B. et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med 3(7), 524–33 (2015).
Accurso, F. J. et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med 363(21), 1991–2003 (2010).
Ren, C. L. et al. Cystic Fibrosis Foundation Pulmonary Guidelines. Use of Cystic Fibrosis Transmembrane Conductance Regulator Modulator Therapy in Patients with Cystic Fibrosis. Ann Am Thorac Soc 15(3), 271–80 (2018).
Patel, S. et al. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database Syst Rev 3, CD009841 (2015).
Aslam, A. A., Higgins, C., Sinha, I. P. & Southern, K. W. Ataluren and similar compounds (specific therapies for premature termination codon class I mutations) for cystic fibrosis. Cochrane Database Syst Rev 1, CD012040 (2017).
Whiting, P. et al. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis. Health Technol Assess 18(18), 1–106 (2014).
Rowe, S. M. et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med 377(21), 2024–35 (2017).
Marigowda, G., Liu, F. & Waltz, D. Effect of bronchodilators in healthy individuals receiving lumacaftor/ivacaftor combination therapy. J Cyst Fibros 16(2), 246–9 (2017).
McKone, E. F. et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir Med 2(11), 902–10 (2014).
Konstan, M. W. et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir Med 5(2), 107–18 (2017).
Davies, J. C. et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med 4(2), 107–15 (2016).
Rosenfeld M, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir Med (2018).
Jennings, M. T. et al. An Observational Study of Outcomes and Tolerances in Patients with Cystic Fibrosis Initiated on Lumacaftor/Ivacaftor. Ann Am Thorac Soc 14(11), 1662–6 (2017).
Hubert, D. et al. Real-life initiation of lumacaftor/ivacaftor combination in adults with cystic fibrosis homozygous for the Phe508del CFTR mutation and severe lung disease. J Cyst Fibros 16(3), 388–91 (2017).
Clancy, J. P. et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 67(1), 12–8 (2012).
Moher, D., Liberati, A., Tetzlaff, J., Altman, D. G. & Group, P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA Statement. Open Med 3(3), e123–30 (2009).
Elborn, J. S. et al. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: a pooled analysis. Lancet Respir Med 4(8), 617–26 (2016).
Acknowledgements
B.S.Q. receives salary support from a Michael Smith Foundation for Health Research Scholar Award and CF Canada Clinician-Scientist Award. No grants or third-party funding was provided for this study.
Author information
Authors and Affiliations
Contributions
A.H., M.K., B.S.Q. contributed to all aspects of this study including study concept and design, conducting the literature search, study design, data collection, data analysis, data interpretation and writing of the manuscript. S.D., C.L.Y., K.S. contributed to data analysis, data interpretation, and writing of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
B.S.Q. has received consulting fees from Proteostasis Therapeutics Inc. and Horizon Pharma and has served as site PI for Vertex sponsored clinical trials.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Habib, AR.R., Kajbafzadeh, M., Desai, S. et al. A Systematic Review of the Clinical Efficacy and Safety of CFTR Modulators in Cystic Fibrosis. Sci Rep 9, 7234 (2019). https://doi.org/10.1038/s41598-019-43652-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43652-2
This article is cited by
-
Clinical significance of BPI-ANCA in patients with cystic fibrosis: a single center prospective study
Scientific Reports (2023)
-
Role of inflammation and oxidative stress in tissue damage associated with cystic fibrosis: CAPE as a future therapeutic strategy
Molecular and Cellular Biochemistry (2022)
-
The NALCN channel regulates metastasis and nonmalignant cell dissemination
Nature Genetics (2022)
-
The impact of physical activity and exercise interventions for physical health in people with cystic fibrosis: protocol for a systematic review
Systematic Reviews (2021)
-
The effect of CFTR modulators on a cystic fibrosis patient presenting with recurrent pancreatitis in the absence of respiratory symptoms: a case report
BMC Gastroenterology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
