
Abstract
Standard-dose intravenous recombinant interleukin-2 (rIL-2) is indicated for the treatment of some subtypes of cancer; however, severe adverse events, including venous thromboembolism (VTE), may complicate its administration. Low-dose subcutaneous rIL-2 is being studied for the management of immune-mediated diseases, since it can modulate the immunological response by specifically targeting T regulatory (Treg) cells; importantly, it is supposed to cause fewer or no complications. In this systematic review and meta-analysis of phase II-III randomized controlled trials (RCTs), we investigated the safety of low-dose (<6 Million International Unit [MIU]/day) and ultra-low-dose (≤1 MIU/day) rIL-2 for severe adverse events (grade III-V) with a focus on VTE. Data of 1,321 patients from 24 RCTs were analysed: 661 patients were randomized to the rIL-2 arm (on top of standard of care) and 660 patients to standard of care alone or placebo. Two studies reported higher rates of thrombocytopenia in the low-dose rIL-2 arm. Ultra-low-dose rIL-2 was reported to be well tolerated in 6 studies with a negligible rate of severe adverse events. Symptomatic VTE events were not reported in any of the study arms (absolute risk difference 0% [95%CI −0.1%; +0.1%]). Our results may facilitate the study and introduction in clinical practice of low-dose rIL-2 for potentially new indications.
Similar content being viewed by others


Introduction
Interleukin 2 (IL-2) is required for the activation, growth, and differentiation of several families of immune cells, including T lymphocytes, B lymphocytes, and natural killer (NK) cells1,2. Human recombinant IL-2 (rIL-2), the highly purified protein used in clinical practice, has the same biological activity of the native molecule3. Current main indications of rIL-2 include the treatment of metastatic renal cell carcinoma and melanoma4,5, for which high-dose intravenous regimens (e.g. above 50 Million International Unit [MIU]/8 hours) are recommended6.
In recent years, it has become clear that the immunological effects of rIL-2 are dose dependent. The subcutaneous administration of low- or ultra-low-dose rIL-2 (e.g below 5.4 MIU every 8 hours, or a cumulative dose below 52.5 MIU) exerts distinctive immunological effects in vivo, which may benefit not only the cancer patients for which high-dose rIL2 is already indicated, but also selected groups of patients with other conditions. In fact, low- and ultra-low dose rIL-2 affect the maintenance and proliferation of functional T regulatory cells (Treg) with apparently positive effects on the course of immune-mediated diseases, such as type I diabetes7, systemic lupus erythematosus8,9, immune thrombocytopenia10, vasculitis induced by the hepatitis C virus (HCV)11, and other autoimmune diseases12. Moreover, regulatory T cells, which are reliant on IL-2 levels, might positively influence the process of thrombus resolution after acute venous thromboembolism (VTE)13,14.
Serious safety concerns have been raised around the use of high-dose IL-2, which has been reported to potentially cause acute thromboembolic events, including VTE15,16, as well as cardiac, cerebral, and hepatic venous thrombosis17,18,19, as also reported in the product monograph20. In the Evaluation of Subcutaneous Proleukin in a Randomized International Trial (ESPRIT) study, the hazard ratio for arterial and venous events with IL-2 administered on top of antiretroviral therapy (vs. antiretroviral therapy alone) was 2.80 (95% Confidence Interval [95% CI] 1.53–5.15) with four-month rates of 8.6% and 3.7%, respectively16. The most frequent type of event was deep vein thrombosis, occurring in 10 (2.1%) and 2 (0.5%) patients, respectively16. In a pathophysiological perspective, such increased risk in thrombotic events is explained by the fact that IL-2 can increase platelet adherence21 and activate the intrinsic coagulation pathway22. Furthermore, animal models suggest that rIL-2 inhibits the expression and activity cytochromes and transporters involved in the absorption and metabolism of oral anticoagulants23,24. Other severe adverse events (AE) reported in patients administered high-dose rIL-2 include capillary leak syndrome, sepsis, and autoimmune reactions20.
In contrast, preliminary findings suggest that low-dose rIL-2 may be better tolerated and characterized by lower AE rates compared with higher doses25. However, in the absence of any comprehensive assessment of its safety, the administration of rIL-2 for research purposes to patients diagnosed with conditions characterized by an intrinsic high risk of severe adverse events and venous thromboembolism should be cautious26. In our systematic review and meta-analysis, we investigated the safety of low- or ultra-low-dose subcutaneous rIL-2 administration in humans.
Results
In our systematic review, we searched the literature and meta-analysed the results of phase II-III randomized controlled trials in which patients were assigned to receive either rIL-2 on top of the standard of care or standard of care alone.
Study selection
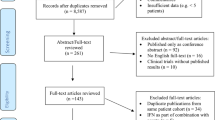
Following the predefined search strategy and after removal of duplicated records, our literature search identified 1,672 records. A total of 24 articles were selected, after the full-text evaluation27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50. Supplementary Fig. S1 summarizes the process of study selection and the reasons for study exclusion. The search for unpublished randomized clinical trials on the topic resulted in the evaluation of 79 additional studies that were registered on the database (http://www.clinicaltrials.gov): however, none of them met the inclusion criteria, since they were single-arm studies (n = 45), non-randomized (n = 10), used a high dose of rIL-2 (n = 11), studied another exposure (n = 4), or already published and included in our study (n = 9).
Characteristics of the included studies and quality assessment
The sample size of the 24 included studies ranged from 10 to 241 patients for a total number of 1,321 patients enrolled27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50: of these, 661 were randomized to receive low-dose rIL-2 (on top of the standard of care) and 660 the standard of care alone or the placebo. One trial included patients with autoimmune disease (Type 1 diabetes)33, eight trials focused on patients with active cancer (usually at stage II or higher)36,38,40,41,42,43,44,49, and 15 trials enrolled patients with infectious diseases, more often by human immunodeficiency virus (HIV) (n = 11)27,28,31,32,35,37,39,45,47,48,50. The average follow-up time ranged from 7 to 973 days.
Although the therapeutic dose of rIL-2 for the approved indications (renal carcinoma and melanoma) is calculated based on weight (kg) or body surface (m2), 19 of the 24 included trials in our review used a fixed low dose of rIL-2, ranging from 0.33 MIU/day to 6 MIU/day. The highest daily rIL-2 dose (6 MIU/day) was administered in three trials enrolling HIV positive patients28,32,50 and in one trial on melanoma patients41. Four trials administered various dosages of rIL-228,29,30,31. The detailed general characteristics of the included studies are summarised in Table 1.
The assessment of quality and risk of bias showed that three studies were classified to a low risk of bias33,34,43. Risk of selection bias was not detected in only one of the included studies, due to the manner in which participant were screened for eligibility and enrolled47. Since most of the included studies were open label trials, the lack of blinding of participants and personnel carried a high or unclear risk of bias; however, blinding of outcome was properly conducted in more than 50% of studies. Risk of incomplete outcome was assessed as low in around 50% of the included studies while we evaluated the risk of selective reporting bias as high in 50% of included studies (Table 2 and Supplementary Fig. S2).
Safety of rIL-2
Of the 24 included studies, 14 (58%) reported no grade III-V AEs in either the intervention or the control arm27,28,29,30,33,34,36,39,40,41,42,45,46,49. In eight studies, no statistically significant difference was observed between the intervention and comparator arms32,35,37,38,43,47,48,50. None of the included trials reported any major bleeding complications. Two studies reported higher rates of thrombocytopenia among rIL-2-treated patients. In one of these studies, that was conducted in HIV patients, thrombocytopenia was reported only in the rIL-2 arm31; in the other study, that was conducted in lung cancer patients, it was reported in both arms, with a statistically significantly higher rate in the rIL-2 arm44. The summary of grade III-V AEs is presented in Table 3.
A total of six studies investigated the effects of an ultra-low-dose of rIL-2 (≤1 MIU/day)27,33,34,36,46,48. Of these, two did not report any AE27,36, which were reported in long-term follow-up studies to occur in similar proportions in rIL-2 and placebo or comparator. Local reactions at the injection sites were the most frequently reported grade I-II AE in two studies, with a prevalence of 50% to 100% among patients with type I diabetes and tuberculosis, respectively33,34. Other reactions included influenza-like syndrome, headache, and dyspnoea (Supplementary Table S1).
None of the included interventional studies reported any thromboembolic complications. Therefore, the pooled absolute risk difference between the rIL-2 arm and controls essentially was 0 (95% CI: −0.1%; +0.1%) (Supplementary Fig. S3). In order to minimize the risk of underreporting, we contacted all 17 corresponding authors of the included research articles for whom the e-mail address was available27,28,31,32,33,34,36,37,38,39,41,43,44,46,47,48,49: seven of them were able to retrospectively review the study case report forms and data, and confirmed that no thromboembolic events were observed (total number of 626 patients from those studies)34,39,41,43,44,47,49. Ten authors did not answer to our e-mails or their e-mail address was no longer active.
Discussion
Our systematic review and meta-analysis of the results of phase II-III clinical trials showed that the use of subcutaneous low-dose rIL-2 on top of the standard of care is well tolerated and does not appear to increase the risk of developing thromboembolic events in patients with various conditions, notably HIV, tuberculosis, autoimmune diseases, or cancer. The safety profile of rIL-2 appeared particularly favourable when rIL-2 was given at ultra-low dose. Two trials reported a possible association of rIL-2 with thrombocytopenia after multiple cycles of treatment in populations at an intrinsic higher risk for this complication, such as cancer or HIV patients.
Since rIL-2 can selectively expand T-lymphocytes populations, it had been tested experimentally in a number of chronic diseases, including chronic infections or cancers, in which T-cell activation plays a pathophysiological and possibly therapeutic role. The initial lack of positive findings prompted researchers to progressively increase doses and, in this perspective, a number of interventional studies were conducted more than two decades ago. However, this approach led to higher rate and severity of AE with no significant gains in terms of efficacy51. Recently, it has been observed that this might have been a misleading approach: high doses of rIL-2 are more than sufficient to saturate both the high- and the intermediate-affinity rIL-2 receptor, but they also lead to massive release of proinflammatory cytokines and directly trigger the capillary-leak syndrome52. In contrast, the ultra-low doses of rIL-2 seem sufficient to trigger Treg lymphocytes expansion, probably because of its affinity for the rIL-2 heterotrimeric receptor components, with higher doses providing no added value53,54. This cell expansion might be clinically relevant: a recent human study suggested that administration of low-dose rIL-2 to patients with active systemic lupus erythematosus changes the proportions of T effector memory cells (Teffector) and Treg, which in turn reduces disease activity55; and animal studies have shown that a selective expansion and activation of Treg lymphocytes as achieved by ultra-low rIL-2 may influence thrombus resolution13. In this perspective, our finding that ultra-low-dose rIL-2 is safe appears promising with a view to future studies in patients with or at risk for VTE.
Although we aggregated all the available data on the safety of low-dose rIL-2, the limited number of individual patients in our analyses should be acknowledged, which may hinder the clinical interpretation of the results. This is inevitable because of the novelty of the low-dose concept of rIL-2 and because the proposed indications include relatively rare diseases. In our systematic review, selective reporting was the most prominent source of bias. We focused on AEs rather than on efficacy outcomes of the trials; venous thrombotic events, in particular, have been shown to be often underreported in randomized trials56. Accordingly, we attempted to minimize this risk by directly contacting all the authors of the studies included in our systematic review and asking for confirmation that no event of interest had occurred. It must be noted that we did not meta-analyse the overall rates of AEs, as studies varied substantially in the nature and severity of the diseases studied as well as in the assessment of AEs, thus preventing any meaningful interpretation of pooled results. The extracted data on all AEs showed that thrombocytopenia was the only severe AE (grade III or higher) reported with significantly higher rates in low-dose rIL-2 than controls in HIV patients: 4% vs 0%, and in lung cancer patients: 25.5% vs. 9.9%31,44. Both studies were conducted on patient populations with a very high baseline risk for thrombocytopenia due to the primary diseases, especially in the case of HIV57. This specific AE was not observed in studies using ultra-low-dose rIL-2 (Supplementary Table S1)27,33,34,36,46,48. Until further evidence in this regard is available, regular monitoring of blood counts should be considered when rIL-2 administration is planned in patients at a higher risk for thrombocytopenia.
In conclusion, the use of low- or ultra-low-dose subcutaneous rIL-2 did not appear to be associated with an increased risk of venous thromboembolic events in randomized controlled trials. Administration of low-dose rIL-2 can be considered safe for clinical and experimental use in humans since an overall low rate of severe adverse events was observed, especially if given at ultra-low dose. It is, however, reasonable to monitor platelet count in patients at risk for thrombocytopenia, e.g. in the presence of active cancer or HIV, in particular if multiple treatment cycles with rIL-2 are envisaged.
Methods
Study selection
The systematic search was conducted in MEDLINE (via PubMed), Google Scholar, the database of ‘http://www.clinicaltrials.gov’, and in the Cochrane Collaboration database, from inception to April 7th, 2018, without any language restrictions. The full search strategy is available as Supplementary Material. We complemented this search by manually reviewing the references of retrieved articles, relevant review papers, guidelines documents, and the grey literature. Authors of the selected studies were contacted by electronic mail if there was ambiguity about original data and to receive confirmation regarding the methods of AE assessment, as well as the characteristics of reported AEs. After removal of duplicates, titles and abstracts of the articles were screened independently by two reviewers (SHM and MJ) for eligibility; disagreements were solved by a third reviewer (SB). The present review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement methodology58.
Search strategy
We included peer-reviewed studies meeting the following criteria:
-
Study population composed of adults enrolled into a randomized controlled phase II-III trial investigating the effects of rIL-2 on surrogate laboratory parameters, on clinical outcomes, or both, and administered for any of the following indications: (i) viral or bacterial infection, (ii) solid cancer, or (iii) active autoimmune disease.
-
Intervention: subcutaneous rIL-2 given at a low dose on top of the standard of care. Low-dose rIL-2 was defined by a daily total dose not exceeding 6 MIU and/or a cumulative dose not exceeding 60 MIU according to the definition of low-dose rIL-2 provided by Klatzmann and Abbas25.
-
Comparator: standard of care alone (or placebo).
-
Primary outcomes: AE (grading according to the Common Terminology Criteria for Adverse Events) which include acute VTE, defined as objectively diagnosed symptomatic deep vein thrombosis, pulmonary embolism or other major thromboses (cerebral vein thrombosis, splanchnic thrombosis, central catheter thrombosis) as well as major bleeding59;
-
Length of follow-up: as specified in the original article. If multiple cycles were administered, the primary focus was put on the first rIL-2 treatment cycle.
-
Study design: phase II or phase III randomized controlled trials.
Data extraction
Full-texts of all included studies were retrieved through the library of the Johannes Gutenberg University Mainz or by contacting the authors. The following information was extracted from the included studies: year of publication, patients’ baseline characteristics, sample size, duration of follow-up, disease for which patients were treated, procedure for recording of the AEs, interventions including the administered dose, rate of study outcomes. We extracted data regarding severe AEs (e.g. grade III or higher) which include major bleeding events. We furthermore focused on thromboembolic events. We predefined a subgroup analysis focusing on studies adopting an ultra-low-dose rIL-2 (≤1 MIU/day).
Quality assessment
Two reviewers (MJ and SB) independently assessed the quality and the risk of bias in accordance to the criteria recommended by the Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.060. Additional details regarding this process are provided as Supplementary Material. Furthermore, we assessed specifically the risk of bias for evaluation and reporting of AE as recommended by the Cochrane Adverse Effects Methods Group60,61. The presence of the following items was evaluated: description of the method used for the assessment of AEs, exclusion of patients from the AE analysis, and presence of numerical data by intervention group.
Statistical analysis
The number of subjects who developed the outcome of interest was extracted; if no case was recorded, the authors were contacted for confirmation. We relied on data from the original articles when the authors did not reply. Risk differences and 95% CI for developing the VTE were calculated for all studies separately and subsequently pooled using the Mantel-Haenszel random effects model. Heterogeneity of results among studies was tested with the I2 measure, which describes the percentage of total variation across studies that is due to heterogeneity rather than chance (I2 values > 50% indicate a substantial level of heterogeneity). Review Manager was used to pool the data (RevMan; version 5.3 for Windows; Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014).
Data Availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information Files).
References
Watson, J., Mochizuki, D. & Gillis, S. T-cell growth factors: interleukin 2. Immunol Today 1, 113–117, https://doi.org/10.1016/0167-5699(80)90047-X (1980).
Gaffen, S. L. & Liu, K. D. Overview of interleukin-2 function, production and clinical applications. Cytokine 28, 109–123, https://doi.org/10.1016/j.cyto.2004.06.010 (2004).
Doyle, M. V., Lee, M. T. & Fong, S. Comparison of the biological activities of human recombinant interleukin-2(125) and native interleukin-2. J Biol Response Mod 4, 96–109 (1985).
Hotte, S., Waldron, T., Canil, C. & Winquist, E. Interleukin-2 in the treatment of unresectable or metastatic renal cell cancer: a systematic review and practice guideline. Can Urol Assoc J 1, 27–38 (2007).
Petrella, T. et al. Single-agent interleukin-2 in the treatment of metastatic melanoma: a systematic review. Cancer Treat Rev 33, 484–496, https://doi.org/10.1016/j.ctrv.2007.04.003 (2007).
Rosenberg, S. A. IL-2: the first effective immunotherapy for human cancer. J Immunol 192, 5451–5458, https://doi.org/10.4049/jimmunol.1490019 (2014).
Hulme, M. A., Wasserfall, C. H., Atkinson, M. A. & Brusko, T. M. Central role for interleukin-2 in type 1 diabetes. Diabetes 61, 14–22, https://doi.org/10.2337/db11-1213 (2012).
Neely, J. & von Scheven, E. Autoimmune haemolytic anaemia and autoimmune thrombocytopenia in childhood-onset systemic lupus erythematosus: updates on pathogenesis and treatment. Curr Opin Rheumatol 30, 498–505, https://doi.org/10.1097/BOR.0000000000000523 (2018).
von Spee-Mayer, C. et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Annals of the rheumatic diseases 75, 1407–1415, https://doi.org/10.1136/annrheumdis-2015-207776 (2016).
Zhang, J. et al. Therapeutic potential of low-dose IL-2 in immune thrombocytopenia: An analysis of 3 cases. Cytometry B Clin Cytom 94, 428–433, https://doi.org/10.1002/cyto.b.21601 (2018).
Saadoun, D. et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med 365, 2067–2077, https://doi.org/10.1056/NEJMoa1105143 (2011).
Rosenzwajg, M. et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Annals of the rheumatic diseases, https://doi.org/10.1136/annrheumdis-2018-214229 (2018).
Luther, N. et al. Innate Effector-Memory T-Cell Activation Regulates Post-Thrombotic Vein Wall Inflammation and Thrombus Resolution. Circ Res 119, 1286–1295, https://doi.org/10.1161/CIRCRESAHA.116.309301 (2016).
Prochaska, J. H. et al. Acute deep vein thrombosis suppresses peripheral T cell effector function. Br J Haematol, https://doi.org/10.1111/bjh.15192 (2018).
Amato, R. J., Morgan, M. & Rawat, A. Phase I/II study of thalidomide in combination with interleukin-2 in patients with metastatic renal cell carcinoma. Cancer 106, 1498–1506, https://doi.org/10.1002/cncr.21737 (2006).
Group, I.-E. S. et al. Interleukin-2 therapy in patients with HIV infection. N Engl J Med 361, 1548–1559, https://doi.org/10.1056/NEJMoa0903175 (2009).
Junghans, R. P., Manning, W., Safar, M. & Quist, W. Biventricular cardiac thrombosis during interleukin-2 infusion. N Engl J Med 344, 859–860, https://doi.org/10.1056/nejm200103153441118 (2001).
Hotton, K. M. et al. A phase Ib/II trial of granulocyte-macrophage-colony stimulating factor and interleukin-2 for renal cell carcinoma patients with pulmonary metastases: a case of fatal central nervous system thrombosis. Cancer 88, 1892–1901 (2000).
Nagafuchi, S. et al. Budd-Chiari syndrome and Epstein-Barr virus (EBV) associated plasmacytoma in a patient with chronic active EBV infection. The Clinical investigator 72, 883–886 (1994).
Novartis Pharmaceuticals Canada Inc. PRODUCT MONOGRAPH PROLEUKIN (aldesleukin) Interleukin-2 Lyophilized powder 22 million IU/vial. (2014).
Edwards, M. J. et al. Interleukin 2 acutely induces platelet and neutrophil-endothelial adherence and macromolecular leakage. Cancer research 52, 3425–3431 (1992).
Fleischmann, J. D., Shingleton, W. B., Gallagher, C., Ratnoff, O. D. & Chahine, A. Fibrinolysis, thrombocytopenia, and coagulation abnormalities complicating high-dose interleukin-2 immunotherapy. The Journal of laboratory and clinical medicine 117, 76–82 (1991).
Hosten, B. et al. Interleukin-2 treatment effect on imatinib pharmacokinetic, P-gp and BCRP expression in mice. Anticancer Drugs 21, 193–201, https://doi.org/10.1097/CAD.0b013e3283349913 (2010).
Di Minno, A. et al. Old and new oral anticoagulants: Food, herbal medicines and drug interactions. Blood Rev 31, 193–203, https://doi.org/10.1016/j.blre.2017.02.001 (2017).
Klatzmann, D. & Abbas, A. K. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol 15, 283–294, https://doi.org/10.1038/nri3823 (2015).
Kearon, C. et al. Categorization of patients as having provoked or unprovoked venous thromboembolism: guidance from the SSC of ISTH. J Thromb Haemost 14, 1480–1483, https://doi.org/10.1111/jth.13336 (2016).
Amendola, A. et al. Decreased CD95 expression on naive T cells from HIV-infected persons undergoing highly active anti-retroviral therapy (HAART) and the influence of IL-2 low dose administration. Irhan Study Group. Clinical and experimental immunology 120, 324–332 (2000).
Arno, A. et al. Efficacy of low-dose subcutaneous interleukin-2 to treat advanced human immunodeficiency virus type 1 in persons with </=250/microL CD4 T cells and undetectable plasma virus load. J Infect Dis 180, 56–60, https://doi.org/10.1086/314831 (1999).
Artillo, S. et al. Double-blind, randomized controlled trial of interleukin-2 treatment of chronic hepatitis B. J Med Virol 54, 167–172 (1998).
Bruch, H. R. et al. Treatment of Chronic Hepatitis-B with Interferon-Alpha-2b and Interleukin-2. Journal of hepatology 17, S52–S55, https://doi.org/10.1016/S0168-8278(05)80424-6 (1993).
Carr, A. et al. Outpatient continuous intravenous interleukin-2 or subcutaneous, polyethylene glycol-modified interleukin-2 in human immunodeficiency virus-infected patients: a randomized, controlled, multicenter study. Australian IL-2 Study Group. J Infect Dis 178, 992–999 (1998).
De Paoli, P. et al. Changes in thymic function in HIV-positive patients treated with highly active antiretroviral therapy and interleukin-2. Clin Exp Immunol 125, 440–446 (2001).
Hartemann, A. et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 1, 295–305, https://doi.org/10.1016/S2213-8587(13)70113-X (2013).
Johnson, J. L. et al. Randomized trial of adjunctive interleukin-2 in adults with pulmonary tuberculosis. Am J Respir Crit Care Med 168, 185–191, https://doi.org/10.1164/rccm.200211-1359OC (2003).
Lalezari, J. P. et al. Low-dose daily subcutaneous interleukin-2 in combination with highly active antiretroviral therapy in HIV+ patients: a randomized controlled trial. HIV Clin Trials 1, 1–15, https://doi.org/10.1310/T5FR-8JPX-0NEF-XDKD (2000).
Li, Y. et al. Interleukin-2 administration after modified radical mastectomy in breast cancer therapy increases peripheral regulatory T cells. Int J Clin Exp Med 8, 7816–7822 (2015).
Losso, M. H. et al. A randomized, controlled, phase II trial comparing escalating doses of subcutaneous interleukin-2 plus antiretrovirals versus antiretrovirals alone in human immunodeficiency virus-infected patients with CD4+ cell counts >/=350/mm3. J Infect Dis 181, 1614–1621, https://doi.org/10.1086/315430 (2000).
Mantovani, G. et al. Neo-adjuvant chemo-(immuno-)therapy of advanced squamous-cell head and neck carcinoma: a multicenter, phase III, randomized study comparing cisplatin +5-fluorouracil (5-FU) with cisplatin +5-FU + recombinant interleukin 2. Cancer Immunol Immunother 47, 149–156 (1998).
Marchetti, G. et al. Low-dose prolonged intermittent interleukin-2 adjuvant therapy: Results of a randomized trial among human immunodeficiency virus-positive patients with advanced immune impairment. Journal of Infectious Diseases 186, 606–616, https://doi.org/10.1086/342479 (2002).
Nichols, P. H. et al. Peri-operative modulation of cellular immunity in patients with colorectal cancer. Clinical and experimental immunology 94, 4–10 (1993).
Nicholson, S. et al. A randomized phase II trial of SRL172 (Mycobacterium vaccae)+/− low-dose interleukin-2 in the treatment of metastatic malignant melanoma. Melanoma Res 13, 389–393, https://doi.org/10.1097/01.cmr.0000056252.56735.1a (2003).
Perillo, A. et al. Administration of low-dose interleukin-2 plus G-CSF/EPO early after autologous PBSC transplantation: effects on immune recovery and NK activity in a prospective study in women with breast and ovarian cancer. Bone Marrow Transplant 30, 571–578, https://doi.org/10.1038/sj.bmt.1703687 (2002).
Procopio, G. et al. Sorafenib with interleukin-2 vs sorafenib alone in metastatic renal cell carcinoma: the ROSORC trial. Br J Cancer 104, 1256–1261, https://doi.org/10.1038/bjc.2011.103 (2011).
Ridolfi, L. et al. Chemotherapy with or without low-dose interleukin-2 in advanced non-small cell lung cancer: results from a phase III randomized multicentric trial. Int J Oncol 39, 1011–1017, https://doi.org/10.3892/ijo.2011.1099 (2011).
Ruxrungtham, K. et al. A randomized, controlled 24-week study of intermittent subcutaneous interleukin-2 in HIV-1 infected patients in Thailand. AIDS 14, 2509–2513 (2000).
Shen, H. et al. The beneficial effects of adjunctive recombinant human interleukin-2 for multidrug resistant tuberculosis. Arch Med Sci 11, 584–590, https://doi.org/10.5114/aoms.2015.52362 (2015).
Smith, K. A. et al. Immunotherapy with canarypox vaccine and interleukin-2 for HIV-1 infection: termination of a randomized trial. PLoS Clin Trials 2, e5, https://doi.org/10.1371/journal.pctr.0020005 (2007).
Vogler, M. A. et al. Daily low-dose subcutaneous interleukin-2 added to single- or dual-nucleoside therapy in HIV infection does not protect against CD4(+) T-cell decline or improve other indices of immune function: Results of a randomized controlled clinical trial (ACTG 248). Jaids-J Acq Imm Def 36, 576–587, https://doi.org/10.1097/00126334-200405010-00005 (2004).
Woodson, E. M. et al. Assessment of the toxicities of systemic low-dose interleukin-2 administered in conjunction with a melanoma peptide vaccine. J Immunother 27, 380–388 (2004).
Zanussi, S. et al. Immunological changes in peripheral blood and in lymphoid tissue after treatment of HIV-infected subjects with highly active anti-retroviral therapy (HAART) or HAART + IL-2. Clinical and experimental immunology 116, 486–492 (1999).
Rosenberg, S. A. Interleukin-2 and the development of immunotherapy for the treatment of patients with cancer. The cancer journal from Scientific American 6(Suppl 1), S2–7 (2000).
Waldmann, T. A. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: implications for cancer therapy. Cancer immunology research 3, 219–227, https://doi.org/10.1158/2326-6066.Cir-15-0009 (2015).
Ito, S. et al. Ultra-low dose interleukin-2 promotes immune-modulating function of regulatory T cells and natural killer cells in healthy volunteers. Molecular therapy: the journal of the American Society of Gene Therapy 22, 1388–1395, https://doi.org/10.1038/mt.2014.50 (2014).
Tang, Q. Therapeutic Window of Interleukin-2 for Autoimmune Diseases. 64, 1912–1913, https://doi.org/10.2337/db15-0188%J Diabetes (2015).
He, J. et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nature medicine 22, 991–993, https://doi.org/10.1038/nm.4148 (2016).
Stuijver, D. J. et al. Under-reporting of venous and arterial thrombotic events in randomized clinical trials: a meta-analysis. Intern Emerg Med 10, 219–246, https://doi.org/10.1007/s11739-014-1168-2 (2015).
Franchini, M., Veneri, D. & Lippi, G. Thrombocytopenia and infections. Expert review of hematology 10, 99–106, https://doi.org/10.1080/17474086.2017.1271319 (2017).
Moher, D., Liberati, A., Tetzlaff, J. & Altman, D. G. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med 151, 264–269, w264 (2009).
Schulman, S. & Kearon, C. Subcommittee on Control of Anticoagulation of the, S., Standardization Committee of the International Society on, T. & Haemostasis. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J Thromb Haemost 3, 692–694, https://doi.org/10.1111/j.1538-7836.2005.01204.x (2005).
Higgins, J. P. et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ 343, d5928, https://doi.org/10.1136/bmj.d5928 (2011).
Loke, Y. K., Price, D. & Herxheimer, A. Systematic reviews of adverse effects: framework for a structured approach. BMC medical research methodology 7, 32, https://doi.org/10.1186/1471-2288-7-32 (2007).
Acknowledgements
This work of S.H.M., C.B., L.V., S.K., Q.K. and S.B. is supported by the German Federal Ministry of Education and Research (BMBF 01EO1003 and 01EO1503).
Author information
Authors and Affiliations
Contributions
S.B. and S.H.M. designed the study. S.B., M.J., L.V. and S.H.M. extracted and analysed the data. S.B., M.J. and S.H.M. drafted the manuscript. S.K., C.B., Q.K. and C.E.K. critically apprised and revised the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mahmoudpour, S., Jankowski, M., Valerio, L. et al. Safety of low-dose subcutaneous recombinant interleukin-2: systematic review and meta-analysis of randomized controlled trials. Sci Rep 9, 7145 (2019). https://doi.org/10.1038/s41598-019-43530-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43530-x
This article is cited by
-
Interleukin-2 and regulatory T cells in rheumatic diseases
Nature Reviews Rheumatology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
