
Abstract
Estrogen-related receptor alpha (ERRα), which shares structural similarities with estrogen receptors, is associated with tumor progression in endometrial cancer, but little is known about the detailed underlying mechanism. We investigated whether ERRα, in cooperation with peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), could participate in epithelial-mesenchymal transition (EMT) in endometrial cancer through cancer-stromal interactions. Two endometrial cancer cell lines, Ishikawa and HEC-1A, transfected with ERRα/PGC-1α expression plasmids or silenced for ERRα expression, were co-cultured with telomerase-transformed human endometrial stromal cells (T-HESCs). We found that EMT-associated factors including vimentin, Snail, and zinc finger E-box binding homeobox 1 were upregulated in cancer cells overexpressing ERRα/PGC-1α and that transforming growth factor-beta (TGF-β) was induced in T-HESCs in the same conditions. In contrast, ERRα knockdown suppressed EMT-associated factors in cancer cells and TGF-β in T-HESCs. ERRα/PGC-1α overexpression increased the expression of EMT-associated factors after TGF-β exposure; however, it decreased E-cadherin at protein level. ERRα knockdown suppressed EMT-associated factors in the presence of TGF-β, whereas E-cadherin remained unchanged. Matrigel invasion assays revealed that ERRα knockdown attenuated the stimulation of migration and invasion by TGF-β. These findings suggest that ERRα is a potential target for inhibiting TGF-β-induced EMT through cancer-stromal interactions in endometrial cancer.
Similar content being viewed by others

Introduction
Endometrial cancer is a leading cause of female genital tract malignancies, and its incidence and death rates have been notably increasing. Most patients (75%) are diagnosed when the disease is still confined to the uterus (International Federation of Gynecology and Obstetrics [FIGO] stage I or II), with 5-year overall survival rates ranging from 74% to 91%1. However, for FIGO stage III and IV, the 5-year overall survival is 57–66% and 20–26%, respectively1. Although several treatment options such as surgery, hormonal therapy, radiation therapy, and chemotherapy are effective for localised disease, only limited options remain if the tumor metastasizes to extrauterine regions2. Therefore, it is important to understand the molecular mechanisms underlying the progression of the disease toward invasion and metastasis.
The conversion of early-stage into advanced-stage tumors is associated with activation of epithelial-mesenchymal transition (EMT), which plays a crucial role in cancer invasion and metastasis3,4,5,6. The acquisition of mesenchymal properties through EMT can promote the detachment of cancer cells from the primary tumor and facilitate their subsequent aggressive dissemination, allowing metastasis to occur7. The connection between the loss of E-cadherin-mediated cell-to-cell adhesion in cancer cells and EMT has been established previouly8. EMT involves the induction or functional activation of mesenchymal markers such as vimentin, and a series of EMT-inducing transcription factors, including Snail and zinc finger E-box binding homeobox 1 (ZEB1). The full spectrum of signaling pathways contributing to EMT in cancer cells remains unclear. Genetic and epigenetic alterations that occur in cancer cells during the course of the primary tumor formation render them especially responsive to EMT-inducing heterotypic signals originating in the tumor microenvironment5.
The tumor microenvironment includes the extracellular matrix (collagen and hyaluronic acid) and many secreted soluble factors, such as Wnt, transforming growth factor beta (TGF-β), Hedgehog, epidermal growth factor, hepatocyte growth factor, and cytokines9,10. Among these signals, TGF-β is the most potent inducer of EMT. TGF-β typically has tumor-suppressing activity in normal cells and early-stage cancers through its ability to induce cell cycle arrest and apoptosis. However, it can serve as a positive regulator of tumor progression and metastasis5,11,12. This switch in TGF-β function is known as the “TGF-β paradox” and is intimately linked to the initiation of EMT13.
Estrogen-related receptor alpha (ERRα), one of the orphan nuclear receptors, shows structural similarities with estrogen receptors, but does not bind to natural estrogens14. ERRα is expressed mostly in tissues with high metabolic demand such as the skeletal muscle, kidney, heart, liver, and adipose tissue15, and plays a predominant role in orchestrating mitochondrial biogenesis and cellular energy metabolism (e.g., oxidative phosphorylation, tricarboxylic acid cycle, fatty acid oxidation, ATP synthesis, and aerobic glycolysis)16,17,18. Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a critical regulator of genes controlling many aspects of energy metabolism, is a powerful transcriptional coactivator of ERRα and regulates the expression and activity of ERRα19,20. ERRα has been reported to be elevated in multiple human cancers including breast, colorectal, ovary, and endometrial cancer21,22,23, and its expression is correlated with poor prognosis24,25,26. We previously demonstrated the tumor-promoting action of ERRα and its association with poor prognosis in endometrial cancer27. However, little has been reported on the detailed underlying mechanism. Here, we investigated whether ERRα could activate EMT of cancer cells through cancer-stromal interactions and thereby promote invasion and metastasis in endometrial cancer.
Results
Upregulation of ERRα in cancer cells induces ERRα and TGF-β in stromal cells
We first confirmed that HEC-1A cells showed higher expression of ERRα and PGC-1α than Ishikawa cells (Fig. 1A). To investigate the cancer-stromal interactions through ERRα in endometrial cancer, human endometrial fibroblasts immortalized with human telomerase reverse transcriptase (T-HESCs) were co-cultured with Ishikawa or HEC-1A cells overexpressing ERRα/PGC-1α (Fig. 1B). Quantitative polymerase chain reaction (qPCR) assays revealed increased levels of ERRα in T-HESCs co-cultured with Ishikawa and HEC-1A cells overexpressing ERRα/PGC-1α (P < 0.01, Fig. 1C). TGF-β is an important mediator in cancer-stromal signaling. Our results showed that overexpression of ERRα/PGC-1α in both cancer cell types induced TGF-β expression in T-HESCs (P < 0.01, Fig. 1D).

Effects of ERRα in cancer cells on ERRα and TGF-β in stromal cells. (A) The expression of ERRα and PGC-1α in Ishikawa and HEC-1A cells was examined by qPCR. (B) T-HESCs were co-cultured with Ishikawa or HEC-1A cells that had been transfected with ERRα/PGC-1α expression plasmids or silenced for ERRα expression. The expression of ERRα (C) and TGF-β (D) was measured using qPCR, in T-HESCs co-cultured with cancer cells overexpressing ERRα/PGC-1α. (E) The effect of ERRα knockdown was evaluated by qPCR and western blot analysis. (F) TGF-β expression was examined in T-HESCs co-cultured with cancer cells with ERRα knockdown. Data represent the mean ± standard error of the mean. P values are based on Student’s t-test. **P < 0.01.
We also performed loss-of-function experiments using siRNA (Fig. 1B). After ERRα knockdown was confirmed by qPCR (P < 0.01) and western blot analysis (Fig. 1E), we examined TGF-β levels in T-HESCs co-cultured with Ishikawa or HEC-1A cells. ERRα knockdown significantly suppressed TGF-β expression in T-HESCs (P < 0.01, Fig. 1F).
ERRα/PGC-1α overexpression induces the expression of EMT markers and the motility of cancer cells through cancer-stromal interactions
To elucidate the association between cancer-stromal interactions and ERRα, we next focused on EMT, as TGF-β is a critical modulator of EMT in tumor progression. The expression of vimentin, Snail, and ZEB1 in cancer cells co-cultured with T-HESCs was examined by qPCR. In Ishikawa and HEC-1A cells overexpressing ERRα/PGC-1α, vimentin, Snail, and ZEB1 were upregulated (P < 0.01, Fig. 2A,B). However, in the monolayer culture system, these responses were weak (data not shown). Wound healing assays showed that ERRα/PGC-1α overexpression significantly induced the motility of Ishikawa and HEC-1A cells (P < 0.05 and P < 0.01 in Ishikawa and HEC-1A cells, respectively). Furthermore, co-culture with T-HESCs tended to increase the responses (Fig. 2C,D). In contrast, vimentin and ZEB1 expression was reduced significantly in Ishikawa cells with silencing of ERRα (P < 0.01), although Snail expression did not change (Fig. 2E). Similarly, the expression of vimentin, Snail, and ZEB1 decreased significantly in HEC-1A cells in which ERRα was silenced (P < 0.01, Fig. 2F).

Effects of ERRα on EMT markers and motility of cancer cells through cancer-stromal interactions. The expression of vimentin, Snail, and ZEB1 in Ishikawa (A) and HEC-1A (B) cells co-cultured with T-HESCs, after overexpression of ERRα/PGC-1α, was examined by qPCR. The wound healing assays of Ishikawa (C) and HEC-1A (D) cells co-cultured with T-HESCs, after knockdown of ERRα (n = 3). The expression of vimentin, Snail, and ZEB1 in Ishikawa (E) and HEC-1A (F) cells co-cultured with T-HESCs, after ERRα knockdown, was examined by qPCR. Data represent the mean ± standard error of the mean. P values are based on Student’s t-test and one-way ANOVA with post-hoc Tukey’s multiple comparison test. *P < 0.05; **P < 0.01.
Upregulation of ERRα is involved in TGF-β-induced EMT
To elucidate the effect of ERRα on TGF-β-induced EMT, we treated Ishikawa and HEC-1A cells overexpressing ERRα/PGC-1α with TGF-β in a monoculture system. Interestingly, ERRα/PGC-1α overexpression significantly induced the expression of vimentin, Snail, and ZEB1 in Ishikawa and HEC-1A cells treated with 10 ng/mL TGF-β (P < 0.01), whereas TGF-β exposure did not have this effect in parental Ishikawa cells (Fig. 3A) and its effect was very limited in parental HEC-1A cells (Fig. 3B). To further confirm this observation, we used western blot analyses to assess the expression of EMT-associated factors. ERRα/PGC-1α overexpression stimulated the expression of vimentin, Snail, and ZEB1 after exposure to TGF-β and reduced the expression of E-cadherin in Ishikawa and HEC-1A cells (Fig. 3C,D). ERRα knockdown reduced the expression of vimentin, Snail, and ZEB1 in the presence of TGF-β. No changes in E-cadherin expression were noted upon ERRα silencing in both cancer cell types (Fig. 3E,F).

Effects of ERRα on TGF-β-induced EMT markers. The expression of ERRα, vimentin, Snail, and ZEB1 was examined in Ishikawa (A) and HEC-1A (B) cells mono-cultured in the presence of 10 ng/mL TGF-β, after overexpression of ERRα/PGC-1α. Western blot analysis of E-cadherin, vimentin, Snail, and ZEB1 in cancer cells treated with TGF-β, after overexpression of ERRα/PGC-1α (C,D). Western blot analysis of these EMT-associated factors in both cancer cell types with ERRα knockdown (E and F). Full-length blots are presented in Supplementary Figs 1–4. Data represent the mean ± standard error of the mean. P values are based on Student’s t-test. **P < 0.01.
ERRα knockdown suppresses TGF-β-induced migration and invasion in endometrial cancer cells
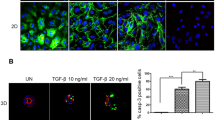
Finally, we evaluated the effect of ERRα on the TGF-β-induced migration and invasion of Ishikawa and HEC-1A cells, using trans-well cell culture and Matrigel invasion chamber systems. We first confirmed that TGF-β exposure significantly induced migration and invasion of Ishikawa and HEC-1A cells (P < 0.01, Fig. 4A,B). We next assessed whether ERRα could regulate migration and invasion in these cells and found that ERRα knockdown attenuated the stimulation of migration and invasion by TGF-β (P < 0.01 and P < 0.05 in Ishikawa and HEC-1A cells, respectively, Fig. 4A,B).

Effects of ERRα knockdown on TGF-β-induced migration and invasion. The migratory and invasive capabilities in Ishikawa and HEC-1A cells treated with TGF-β, after ERRα knockdown, were evaluated using trans-well cell culture and Matrigel invasion chamber systems. Data represent the mean ± standard error of the mean (Ishikawa cells, n = 2; HEC-1A cells, n = 3; A,B). P values are based on one-way ANOVA with post-hoc Tukey’s multiple comparison test. *P < 0.05; **P < 0.01.
Discussion
We elucidated the influence of the tumor microenvironment on the establishment and maintenance of EMT in endometrial cancer cells by co-culturing human endometrial cancer cells and a human endometrial fibroblast cell line. The co-culture system is a useful approach to mimic cancer-stromal interactions. TGF-β plays a key role in tumor progression and metastasis in the tumor microenvironment. Increased expression of TGF-β in various cancers is highly correlated with poor prognosis28,29. However, the function of TGF-β in endometrial cancer remains unclear. Our study focused on determining the effect of TGF-β signaling on the cancer-stromal interactions in endometrial cancer.
First, our results indicated that upregulation of ERRα in endometrial cancer cells induces TGF-β and ERRα expression in stromal cells. A previous study identified soluble factors produced by fibroblasts that stimulate invasion of cancer cells using a co-culture model with breast cancer cell lines and fibroblast cell line established from healthy mammary tissue30. Similarly, in this study, we used an endometrial fibroblast cell line rather than primary fibroblasts isolated from cancer tissues as a non-malignant stromal cell population surrounding the cancer cells. This allowed us to investigate the influence of the altered expression of ERRα in cancer cells on stromal cells. Accumulating evidence suggests that cancer-associated fibroblasts (CAFs) originate from resident tissue fibroblasts31,32,33. CAFs are the main contributors to the association between stromal TGF-β-driven programs and poor clinical outcome in colorectal cancer34. Furthermore, the interaction of colon cancer cells with resident fibroblasts hyperactivates TGF-β signaling in fibroblasts35. Our data indicated that TGF-β signaling in endometrial stromal cells may be mediated by the paracrine ERRα signals of endometrial cancer cells through cancer-stromal interactions.
Second, our results demonstrated that ERRα induces the expression of EMT markers in cancer cells through cancer-stromal interactions. Several studies have described the prognostic impact of the expression of EMT-related factors in endometrial cancer36,37. The increased expression of Snail and ZEB1 is closely related to decreased E-cadherin expression38,39, and this pattern is significantly correlated with myometrial invasion, histological type, and patients’ decreased survival40,41. Our data also revealed that cancer-stromal interactions may promote the motility of cancer cells overexpressing ERRα. Some reports have shown that the interaction between CAFs and cancer cells could enhance the metastatic potential of cancer cells through EMT induced by paracrine TGF-β signaling42,43. Additionally, co-cultures of squamous cell carcinoma cells and stromal fibroblasts revealed that cancer cells moved within tracks in the extracellular matrix produced by the fibroblasts44. In line with these data, our study indicated that stromal cells contribute to EMT in endometrial cancer via secretion of TGF-β.
Finally, our results demonstrated that ERRα promotes the migration and invasion of endometrial cancer cells by enhancing the TGF-β-induced EMT. In support of our results, previous studies have shown that ERRα promotes the migration and invasion of cancer cells by inducing EMT in some kinds of cancers45,46,47,48. Huang X et al. recently reported that ERRα directly binds to the promoter of TGF-β and increases the positive self-feedback regulation of TGF-β in endometrial cancer cells48. Our data indicated that the upregulation of ERRα in cancer cells induces TGF-β expression in stromal cells and the exogenous TGF-β signaling influences EMT in cancer cells. Our findings from the co-culture system suggest that exogenous TGF-β signaling through the cancer-stromal interactions might be more important than endogenous TGF-β from only epithelial cancer cells. Chen Y et al. reported that ERRα could mediate the TGF-β-induced EMT via the transcription of Snail in osteosarcoma cells47. Our study demonstrated that ZEB1 and Snail participate in the ERRα-mediated EMT stimulated by TGF-β in endometrial cancer. Our finding does provide further insight into the potential role of ERRα in progression of endometrial cancer. Furthermore, the level of ERRα in endometrial cancer cells was further increased by positive feedback through TGF-β. In rapidly growing cancers, the relative shortage of blood flow causes ischemia and hypoxia, which increase the expression of ERRα/PGC-1α in cancer cells. We and other investigators have previously demonstrated that ERRα/PGC-1α promotes tumor angiogenesis via induction of VEGF transcription27,49. Therefore, ERRα may be involved in both tumor angiogenesis and tumor invasion in the advanced stages of cancer.
There are several limitations to this study. Although we evaluated EMT in vitro based on downregulation of E-cadherin and upregulation of vimentin, we have not evaluated EMT of cancer cells in vivo. The cancer stroma in vivo has many components other than fibroblasts, such as the extracellular matrix, vascular endothelial cells, immune cells and adipocytes50,51. Moreover, we did not consider how these components function in the tumor microenvironment.
In conclusion, our study revealed for the first time that ERRα plays a crucial role in the TGF-β-induced EMT through cancer-stromal interactions in endometrial cancer cells. Growing evidence indicates targeting TGF-β signaling for cancer treatment is difficult, as TGF-β regulates a broad range of cellular responses, including cell proliferation, differentiation, and apoptosis. Targeting ERRα may be an effective alternative approach for endometrial cancer treatment.
Methods
Cell lines and culture
The human uterine endometrial cancer cell line Ishikawa was provided by the Cell Resource Center for Biomedical Research (Institute of Development, Aging and Cancer, Tohoku University, Sendai, Japan). The human uterine endometrial cancer cell line HEC-1A and the telomerase-transformed human endometrial stromal cells T-HESCs were purchased from the American Type Culture Collection (Manassas, VA, USA). Ishikawa and HEC-1A cells were maintained in Minimum Essential Medium (MEM) (Nacalai Tesque, Kyoto, Japan) with sodium pyruvate, and T-HESCs were cultured in Dulbecco’s Modified Eagle’s Medium/Ham’s F-12 (Nacalai Tesque). Each medium was supplemented with 10% fetal bovine serum (FBS) (Biowest, Nuaille, France) and penicillin-streptomycin (Nacalai Tesque). All cells were cultured at 37 °C in a humidified 5% CO2 atmosphere.
TGF-β was purchased from R&B systems (Minneapolis, MN, USA). Ishikawa and HEC-1A cells were incubated in phenol-red free MEM supplemented with 10 ng/mL TGF-β in a humidified 5% CO2 atmosphere for 48–72 h.
Co-culture system
For physical separation of the endometrial cancer cell lines and T-HESCs, transwell cultures were established in 6- or 12-well plates using Falcon Cell Culture Inserts (pore size 0.4 μm) and Cell Culture Companion Plates (Corning, Corning, NY, USA). First, Ishikawa cells, HEC-1A cells, and T-HESCs were cultured separately until sub-confluency in separate culture plates. T-HESCs were seeded at 1 × 105 cells/mL in Cell Culture Companion Plates, and on the next day, 1 × 105 cells/mL Ishikawa or HEC-1A cells were seeded in the Cell Culture Inserts placed on the top of the T-HESCs. After 24 h of co-culture, Ishikawa or HEC-1A cells and T-HESCs were separated again and examined by qPCR.
Plasmids
The ERRα and PGC-1α expression plasmids were constructed by inserting the full-length human ERRα and PGC-1α genes (NM_004451 and NM_013261) into the pSG5-empty vector (Promega, Madison, WI, USA) to obtain pSG5-ERRα and pSG5-PGC-1α and were kindly provided by Prof. Shiuan Chen. The pSG5-empty plasmid (Promega) was used as a control.
Transient transfection
The transfection experiments were performed on subconfluent cells. Plasmids (pSG5-empty, pSG5-ERRα, and pSG5-PGC-1α) were transfected using Lipofectamine LTX (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. After a 4-h incubation, the transfection medium, containing Lipofectamine LTX and plasmids was removed and replaced with phenol red-free MEM supplemented with 10% dextran-coated charcoal-treated FBS (Biowest) and antibiotics. After a 48-h incubation, the cells were processed for analysis.
RNA interference
The small interfering RNA (siRNA) for ERRα (s4831) and the negative control siRNA (control #1) were Silencer Select siRNAs purchased from Ambion (Austin, TX, USA). The siRNA transfection experiments were performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s protocol. We confirmed the knockdown efficiency of the siRNAs at a final concentration of 10 nM using qPCR and western blotting analysis. The negative control siRNA was used as a control.
RNA extraction and qPCR
RNA extraction and qPCR were performed as previously described52. Total RNA (1 μg) was extracted from cultured cells using the RNeasy Mini kit (QIAGEN, Venlo, Netherlands), and then cDNA was synthesized from 1 μg of RNA using the ReverTra Ace qPCR RT kit (Toyobo, Osaka, Japan). qPCR was performed using the CFX Connect Real-time PCR Detection System (Bio-Rad, Hercules, CA, USA). cDNA samples generated from the total RNA from cells (1 μL) were mixed into 20-μL reactions containing SYBR qPCR Thunderbird master mix (Toyobo) and each primer at 0.3 μM. The following primers were designed with the Primer3Plus free software and purchased from Invitrogen: ERRα 5′-GGCCCTTGCCAATTCAGA-3′ (forward) and 5′-GGCCTCGTGCAGAGCTTCT-3′ (reverse), PGC-1α 5′-TGTGGATGAAGACGGATTGC-3′ (forward) and 5′-GTCAGGCATGGAGGAAGGAC-3′ (reverse), TGF-β 5′-CACTCCCACTCCCTCTCTC-3′ (forward) and 5′-GTCCCCTGTGCCTTGATG-3′ (reverse), E-cadherin 5′-GAGGAGAGcGGTGGTCAAAG-3′ (forward) and 5′-TCCGCCTCCTTCTTCATCAT-3′ (reverse), vimentin 5′-TTGCAGGAGGAGATGCTTCA-3′ (forward) and 5′-GTCAAGACGTGCCAGAGACG-3′ (reverse), Snail 5′-GGCTCCTTCGTCCTTCTCCT-3′ (forward) and 5′-CTGGAGATCCTTGGCCTCAG-3′ (reverse), ZEB1 5′-CACTGCCCAGTTACCCACAA-3′ (forward) and 5′-GGGGAATCAGAATCGTTTGC-3′ (reverse), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) 5′-GCACCGTCAAGGCTGAGAAC-3′ (forward) and 5′-ATGGTGGTGAAGACGCCAGT-3′ (reverse). The mRNA levels of the target genes were quantified using the comparative method (∆∆CT method) and normalized to GAPDH expression.
Wound healing assay
Ishikawa (3.0 × 105) or HEC-1A (3.3 × 105) cells were seeded in the Cell Culture Inserts and T-HESCs (3.0 × 105) were seeded in Cell Culture Companion Plates. On the next day, Ishikawa or HEC-1A cells were wounded with 200-μL pipette tip and placed on the top of T-HESCs or only medium. After a 48-h incubation, the distance migrated was quantitated by acquiring images. Photomicrographs were acquired using a Keyence BZ-X700 microscope (Keyence, Osaka, Japan).
Migration and invasion assay
Migration and invasion assays were performed using uncoated and Matrigel-coated double-chamber systems (pore size 8 μm, Corning BioCoat Matrigel Invasion Chamber, Corning) as previously described52. Ishikawa (4.5 × 104) or HEC-1A (5.0 × 104) cells were seeded into the upper chamber (uncoated or Matrigel-coated inserts) filled with serum-free medium; 10% FBS-containing medium was used as a chemoattractant in the lower chamber. After 24 h, the cells that migrated and invaded into the lower side of the inserts were fixed and stained with the Diff-Quik kit (Sysmex, Kobe, Japan). The number of stained cells was counted in the whole field using an Olympus BX50 microscope (Olympus, Tokyo, Japan) (200×), and photomicrographs were acquired using an Olympus DP22 (Olympus) (100×).
Antibodies
The mouse anti-ERRα (sc-65715), anti-vimentin (sc-66002), and anti-SNAI1 (sc-271977) antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). The mouse anti-E-cadherin antibody (M106) was purchased from Takara Bio Inc. (Shiga, Japan). The rabbit anti-SNAI1 antibody (SAB2108482) was purchased from Sigma-Aldrich (St. Louis, MO, USA). The rabbit anti-ZEB1 antibody (GTX105278) was purchased from Gene Tex (Los Angeles, CA, USA). The rabbit anti-GAPDH (#2118) antibody and the anti-rabbit (#7074) and anti-mouse (#7076) IgG, HRP-linked antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). All antibodies were used at the concentration recommended by the manufacturers.
Western blot analysis
Western blot analysis was performed as previously described52. Cells were washed three times with phosphate-buffered saline and lysed in radioimmunoprecipitation assay (RIPA) buffer (Nacalai Tesque). Cell lysates (10–20 μg) were heated in sodium dodecyl sulfate (SDS) sample buffer (125 mM Tris-HCl, pH 6.8, 4% SDS, 25% glycerol, 10% 2-mercaptoethanol, 0.05 mM phenylmethanesulfonyl fluoride and 0.004% bromophenol blue), separated by 10% e-PAGEL (Atto Corp, Tokyo, Japan) according to the manufacturer’s recommendations, and transferred onto immuno-blot polyvinylidene fluoride (PVDF) membranes (Bio-Rad). The membranes were blocked in PVDF Blocking Reagent for Can Get Signal (Toyobo) for 1 h at 20–25 °C and incubated with the appropriate primary antibody in Can Get Signal Solution 1 (Toyobo) overnight at 4 °C. After washing, the membranes were incubated with the secondary antibody for 1 h at 20–25 °C. The signal was visualized by Chemi-Lumi One (Nacalai Tesque) and analyzed by ChemiDoc XRS + system with Image Lab software (Bio-Rad).
Statistical analysis
Comparisons of the mean and standard error between two groups were made using two-tailed unpaired Student’s t-test. Comparisons of over 3 groups were made using one-way ANOVA with post-hoc Tukey’s multiple comparison test. These statistical analyses were performed using GraphPad Prism ver. 5.04 (GraphPad Software, San Diego, CA, USA). P < 0.05 was considered statistically significant.
Data Availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
References
Creasman, W. T. et al. Carcinoma of the corpus uteri. FIGO 26th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet 95(Suppl 1), S105–143, https://doi.org/10.1016/S0020-7292(06)60031-3 (2006).
Rauh-Hain, J. A. & Del Carmen, M. G. Treatment for advanced and recurrent endometrial carcinoma: combined modalities. Oncologist 15, 852–861, https://doi.org/10.1634/theoncologist.2010-0091 (2010).
Thiery, J. P., Acloque, H., Huang, R. Y. & Nieto, M. A. Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890, https://doi.org/10.1016/j.cell.2009.11.007 (2009).
Yang, J. & Weinberg, R. A. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 14, 818–829, https://doi.org/10.1016/j.devcel.2008.05.009 (2008).
Kalluri, R. & Weinberg, R. A. The basics of epithelial-mesenchymal transition. J Clin Invest 119, 1420–1428, https://doi.org/10.1172/JCI39104 (2009).
Nieto, M. A., Huang, R. Y., Jackson, R. A. & Thiery, J. P. Emt: 2016. Cell 166, 21–45, https://doi.org/10.1016/j.cell.2016.06.028 (2016).
Thiery, J. P. & Sleeman, J. P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 7, 131–142, https://doi.org/10.1038/nrm1835 (2006).
Halbleib, J. M. & Nelson, W. J. Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev 20, 3199–3214, https://doi.org/10.1101/gad.1486806 (2006).
Gavert, N. & Ben-Ze’ev, A. Epithelial-mesenchymal transition and the invasive potential of tumors. Trends Mol Med 14, 199–209, https://doi.org/10.1016/j.molmed.2008.03.004 (2008).
Wu, Y. & Zhou, B. P. New insights of epithelial-mesenchymal transition in cancer metastasis. Acta Biochimica et Biophysica Sinica 40, 643–650, https://doi.org/10.1111/j.1745-7270.2008.00443.x (2008).
Oft, M., Heider, K. H. & Beug, H. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol 8, 1243–1252 (1998).
Bierie, B. & Moses, H. L. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 6, 506–520, https://doi.org/10.1038/nrc1926 (2006).
Morrison, C. D., Parvani, J. G. & Schiemann, W. P. The relevance of the TGF-beta Paradox to EMT-MET programs. Cancer Lett 341, 30–40, https://doi.org/10.1016/j.canlet.2013.02.048 (2013).
Giguere, V., Yang, N., Segui, P. & Evans, R. M. Identification of a new class of steroid hormone receptors. Nature 331, 91–94, https://doi.org/10.1038/331091a0 (1988).
Luo, J. et al. Reduced Fat Mass in Mice Lacking Orphan Nuclear Receptor Estrogen-Related Receptor α. Molecular and Cellular Biology 23, 7947–7956, https://doi.org/10.1128/mcb.23.22.7947-7956.2003 (2003).
Giguere, V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev 29, 677–696, https://doi.org/10.1210/er.2008-0017 (2008).
Cai, Q., Lin, T., Kamarajugadda, S. & Lu, J. Regulation of glycolysis and the Warburg effect by estrogen-related receptors. Oncogene 32, 2079–2086, https://doi.org/10.1038/onc.2012.221 (2012).
Sladek, R., Bader, J. A. & Giguere, V. The orphan nuclear receptor estrogen-related receptor alpha is a transcriptional regulator of the human medium-chain acyl coenzyme A dehydrogenase gene. Mol Cell Biol 17, 5400–5409 (1997).
Willy, P. J. et al. Regulation of PPARgamma coactivator 1alpha (PGC-1alpha) signaling by an estrogen-related receptor alpha (ERRalpha) ligand. Proc Natl Acad Sci USA 101, 8912–8917, https://doi.org/10.1073/pnas.0401420101 (2004).
Schreiber, S. N., Knutti, D., Brogli, K., Uhlmann, T. & Kralli, A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha). J Biol Chem 278, 9013–9018, https://doi.org/10.1074/jbc.M212923200 (2003).
Ariazi, E. A., Kraus, R. J., Farrell, M. L., Jordan, V. C. & Mertz, J. E. Estrogen-related receptor alpha1 transcriptional activities are regulated in part via the ErbB2/HER2 signaling pathway. Mol Cancer Res 5, 71–85, https://doi.org/10.1158/1541-7786.MCR-06-0227 (2007).
Boudjadi, S., Bernatchez, G., Beaulieu, J. F. & Carrier, J. C. Control of the human osteopontin promoter by ERRalpha in colorectal cancer. Am J Pathol 183, 266–276, https://doi.org/10.1016/j.ajpath.2013.03.021 (2013).
Lam, S. S. et al. Targeting estrogen-related receptor alpha inhibits epithelial-to-mesenchymal transition and stem cell properties of ovarian cancer cells. Mol Ther 22, 743–751, https://doi.org/10.1038/mt.2014.1 (2014).
Ariazi, E. A., Clark, G. M. & Mertz, J. E. Estrogen-related receptor alpha and estrogen-related receptor gamma associate with unfavorable and favorable biomarkers, respectively, in human breast cancer. Cancer Res 62, 6510–6518 (2002).
Fujimoto, J. et al. Clinical implication of estrogen-related receptor (ERR) expression in ovarian cancers. J Steroid Biochem Mol Biol 104, 301–304, https://doi.org/10.1016/j.jsbmb.2007.03.016 (2007).
Suzuki, T. et al. Estrogen-related receptor alpha in human breast carcinoma as a potent prognostic factor. Cancer Res 64, 4670–4676, https://doi.org/10.1158/0008-5472.CAN-04-0250 (2004).
Matsushima, H. et al. Anti-tumor effect of estrogen-related receptor alpha knockdown on uterine endometrial cancer. Oncotarget 7, 34131–34148, https://doi.org/10.18632/oncotarget.9151 (2016).
Friess, H. et al. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 105, 1846–1856 (1993).
Chen, C. et al. TGFbeta isoforms and receptors mRNA expression in breast tumours: prognostic value and clinical implications. BMC Cancer 15, 1010, https://doi.org/10.1186/s12885-015-1993-3 (2015).
Olsen, C. J., Moreira, J., Lukanidin, E. M. & Ambartsumian, N. S. Human mammary fibroblasts stimulate invasion of breast cancer cells in a three-dimensional culture and increase stroma development in mouse xenografts. BMC Cancer 10, 444, https://doi.org/10.1186/1471-2407-10-444 (2010).
Shiga, K. et al. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers (Basel) 7, 2443–2458, https://doi.org/10.3390/cancers7040902 (2015).
Orimo, A. et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348, https://doi.org/10.1016/j.cell.2005.02.034 (2005).
Kojima, Y. et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci USA 107, 20009–20014, https://doi.org/10.1073/pnas.1013805107 (2010).
Calon, A. et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 22, 571–584, https://doi.org/10.1016/j.ccr.2012.08.013 (2012).
Hawinkels, L. J. et al. Interaction with colon cancer cells hyperactivates TGF-beta signaling in cancer-associated fibroblasts. Oncogene 33, 97–107, https://doi.org/10.1038/onc.2012.536 (2014).
Koyuncuoglu, M. et al. Tumor budding and E-Cadherin expression in endometrial carcinoma: are they prognostic factors in endometrial cancer? Gynecol Oncol 125, 208–213, https://doi.org/10.1016/j.ygyno.2011.12.433 (2012).
Montserrat, N. et al. Epithelial to mesenchymal transition in early stage endometrioid endometrial carcinoma. Hum Pathol 43, 632–643, https://doi.org/10.1016/j.humpath.2011.06.021 (2012).
Blechschmidt, K. et al. The E-cadherin repressor snail plays a role in tumor progression of endometrioid adenocarcinomas. Diagn Mol Pathol 16, 222–228, https://doi.org/10.1097/PDM.0b013e31806219ae (2007).
Singh, M. et al. ZEB1 expression in type I vs type II endometrial cancers: a marker of aggressive disease. Mod Pathol 21, 912–923, https://doi.org/10.1038/modpathol.2008.82 (2008).
Tanaka, Y. et al. Prognostic impact of EMT (epithelial-mesenchymal-transition)-related protein expression in endometrial cancer. Cancer Biol Ther 14, 13–19, https://doi.org/10.4161/cbt.22625 (2013).
Feng, G., Wang, X., Cao, X., Shen, L. & Zhu, J. ZEB1 expression in endometrial biopsy predicts lymph node metastases in patient with endometrial cancer. Dis Markers 2014, 680361, https://doi.org/10.1155/2014/680361 (2014).
Dimanche-Boitrel, M. T. et al. In vivo and in vitro invasiveness of a rat colon-cancer cell line maintaining E-cadherin expression: an enhancing role of tumor-associated myofibroblasts. Int J Cancer 56, 512–521 (1994).
Soon, P. S. et al. Breast cancer-associated fibroblasts induce epithelial-to-mesenchymal transition in breast cancer cells. Endocr Relat Cancer 20, 1–12, https://doi.org/10.1530/ERC-12-0227 (2013).
Gaggioli, C. et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 9, 1392–1400, https://doi.org/10.1038/ncb1658 (2007).
Huang, J. W. et al. Effects of estrogen-related receptor alpha (ERRalpha) on proliferation and metastasis of human lung cancer A549 cells. J Huazhong Univ Sci Technolog Med Sci 34, 875–881, https://doi.org/10.1007/s11596-014-1367-0 (2014).
Wang, C. W. et al. Aqueous Extract of Paris polyphylla (AEPP) Inhibits Ovarian Cancer via Suppression of Peroxisome Proliferator-Activated Receptor-Gamma Coactivator (PGC)-1alpha. Molecules 21, https://doi.org/10.3390/molecules21060727 (2016).
Chen, Y., Zhang, K., Li, Y. & He, Q. Estrogen-related receptor alpha participates transforming growth factor-beta (TGF-beta) induced epithelial-mesenchymal transition of osteosarcoma cells. Cell Adh Migr 11, 338–346, https://doi.org/10.1080/19336918.2016.1221567 (2017).
Huang, X. et al. Estrogen related receptor alpha triggers the migration and invasion of endometrial cancer cells via up regulation of TGFB1. Cell Adh Migr, 1–10, https://doi.org/10.1080/19336918.2018.1477901 (2018).
Arany, Z. et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 451, 1008–1012, https://doi.org/10.1038/nature06613 (2008).
Liotta, L. A. & Kohn, E. C. The microenvironment of the tumour-host interface. Nature 411, 375–379, https://doi.org/10.1038/35077241 (2001).
Lu, P., Weaver, V. M. & Werb, Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196, 395–406, https://doi.org/10.1083/jcb.201102147 (2012).
Yamamoto, T. et al. Loss of AF-6/afadin induces cell invasion, suppresses the formation of glandular structures and might be a predictive marker of resistance to chemotherapy in endometrial cancer. BMC Cancer 15, 275, https://doi.org/10.1186/s12885-015-1286-x (2015).
Acknowledgements
The authors thank Yunhwa Lee, Ayumi Tanaka, and Ayaka Miura for technical assistance, and Prof. Chen for providing plasmids. This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan [15 K10726, to T.M].
Author information
Authors and Affiliations
Contributions
All authors conceived the study concept and study design. K.Y. and T.M. performed compilation and synthesis of the data, and carried out statistical analyses. J.K. supervised the research project. All authors participated in interpretation of the results and writing of the report, and approved the final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yoriki, K., Mori, T., Kokabu, T. et al. Estrogen-related receptor alpha induces epithelial-mesenchymal transition through cancer-stromal interactions in endometrial cancer. Sci Rep 9, 6697 (2019). https://doi.org/10.1038/s41598-019-43261-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43261-z
This article is cited by
-
Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases
Signal Transduction and Targeted Therapy (2024)
-
ERRα: unraveling its role as a key player in cell migration
Oncogene (2024)
-
Bone marrow mesenchymal stem cells combined with estrogen synergistically promote endometrial regeneration and reverse EMT via Wnt/β-catenin signaling pathway
Reproductive Biology and Endocrinology (2022)
-
Genistein induces long-term expression of progesterone receptor regardless of estrogen receptor status and improves the prognosis of endometrial cancer patients
Scientific Reports (2022)
-
microRNA-26b inhibits growth and cellular invasion of ovarian cancer cells by targeting estrogen receptor α
3 Biotech (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
