
Abstract
RNA interference (RNAi) is a highly specific gene-silencing mechanism that can cause rapid insect mortality when essential genes are targeted. RNAi is being developed as a tool for integrated pest management of some crop pests. Here we focus on an aggressive forest pest that kills extensive tracts of pine forests, the southern pine beetle (SPB), Dendroctonus frontalis. We sought to identify reference genes for quantitative real-time PCR (qPCR) and validate RNAi responses in SPB by mortality and gene silencing analysis. Using an adult beetle feeding bioassay for oral ingestion of dsRNA, we measured the expression and demonstrated knockdown of target genes as well as insect mortality after ingestion of target genes. Our study validates reference genes for expression analyses and demonstrates highly effective RNAi responses in SPB, with RNAi response to some target dsRNAs causing 100% beetle mortality after ingestion.
Similar content being viewed by others

Introduction
The southern pine beetle (SPB), Dendroctonus frontalis, is an oligophagous, tree-killing bark beetle that undergoes extreme population outbreaks that can cause devastating losses in timber1,2 and tourism revenue3, making it the most significant forest pest in the southeastern US4,5. SPB outbreaks, and their management, create significant disturbances, resulting in increased light availability to the forest floor, changes in temperature and soil moisture, competition, and growing space6,7,8,9. The influx of coarse woody debris following tree mortality affects forest succession, nutrient cycling, and wildlife associates7. The loss of large tracts of forest also impacts hydrologic processes that influence water quality and quantity10. In recent decades, changing climate patterns and lack of proactive management have allowed an unprecedented northward range expansion, and SPB is now infesting pine forests of New York, New Jersey, Rhode Island and Connecticut11,12. The alarming geographic range expansion, coupled with persistent outbreaks over its historic range, demonstrate the need for innovative means of managing SPB populations.
RNA interference (RNAi) technology is a novel approach to forest pest management. Introducing exogenous double stranded RNA (dsRNA) into the insect cells activates RNAi pathways that normally function to induce antiviral responses13. The dsRNA is bound by the DICER enzyme, cleaving it into small interfering RNAs (siRNA), which then binds to the RNA induced silencing complex (RISC), where it is digested to produce single-stranded RNA templates able to bind to complementary messenger RNA (mRNA). Binding of siRNA to mRNA induces degradation of the mRNA, preventing its translation and producing nonsensical end products. RNAi technology has proven efficacious in coleopterans, including the notable invasive forest pests, emerald ash borer (Agrilus planipennis)14,15 and Asian longhorned beetle (Anoplophora glabripennis)16. Because the process relies on matching 19 bp or more to target sequences17, RNAi is more target-specific than current insect suppression methods. Non-target effects can be further avoided by choosing novel target genes over highly conserved ones. Though coleopterans appear especially sensitive to RNAi18,19, this sensitivity is variable, and may be influenced by the beetle life stage that is targeted20, the process by which dsRNA is delivered21, and the selected target gene(s)22.
The analysis of target gene silencing using real-time quantitative PCR (qPCR) to evaluate relative gene expression is a standard method used to validate and confirm cellular RNAi machinery. Identifying appropriate reference gene(s) is vital for interpretation of results for other genes that are targeted in experimentation23. Ideal reference genes exhibit expression levels that are conserved across all cells. Reference genes (RG) are essential for proper cell metabolism (e.g. glyceraldehyde-3-phosphate dehydrogenase (GAPDH)) and/or structural integrity (e.g. actin) often exhibit stable expression making them commonly used as reference genes. The stable expression of reference genes is used as an internal control to normalize target gene expression. Though universally present in cells, reference gene expression levels can differ between organisms, and under stress and changing environments; therefore, a gene with constant non-varying expression is preferred and must be identified experimentally by screening.
We sought to validate the RNAi response in SPB by using oral delivery of dsRNA to (i) measure the expression of target genes and demonstrate gene silencing and (ii) evaluate insect mortality after dsRNA ingestion. Due to the lack of known reference genes in SPB, we screened for and identified stable genes for our gene silencing study and future gene expression studies involving SPB. Ours is the first study to validate reference genes for expression analyses and demonstrate RNAi responses in SPB.
Results and Discussion
Reference genes
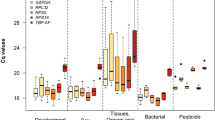
Because qPCR is highly sensitive, normalization by internal controls is essential for accurate quantification of mRNA levels. For our study, moderately expressed genes were chosen as potential reference genes because genes with extremely high or low expression are more likely to introduce variability16. The Cq values for all 8 RG from all four treatments, temperature, photoperiod, sex, and introduction of dsRNA, was between 18 and 26 cycles (Fig. 1A–D). Analysis of the comprehensive values for all four treatments showed succinate dehydrogenase flavoprotein subunit A (sdf) was the least expressed, with average Cq values between 25 and 26, while tubulin (tub) was the most expressed with average Cq values ranging between 18 and 18.5. Ribosomal protein S18 (rps18) was the most moderately expressed RG with values ranging between 19.5 and 20.5 cycles. In this study, we focused primarily on dsRNA treatment beetles, for which sdf was the least expressed, with average values ranging from 22 to 23 cycles, and elongation factor – 1 alpha (ef1a), which was the most highly expressed with Cq values ranging between 13 and 13.5 cycles (Fig. 1D). Ribosomal protein S18 (rps18) was again the most moderately expressed with Cq average values ranging between 19 and 19.5 cycles.

Cq values for 8 candidate reference genes in four independent experiments: (A) temperature, (B) light, (C) sex, (D) RNAi exposure. rps18 was consistently the most moderately expressed. Under RNAi exposure, sdf was the least expressed, and ef1a was the most highly expressed. Bars show the maximum and minimum Cq values.
The Cq values from qPCR analysis were analyzed using four separate algorithms: GeNorm, NormFinder, BestKeeper, and the delta-Ct method. Genes were analyzed by all four algorithms, and comprehensively across the four algorithms, as well as analyzed separately by treatment and comprehensively across all treatments (Table 1). Again, we focused primarily on results from the dsRNA treatments. GeNorm compares all genes based on a gene expression stability value (M). Under dsRNA treatment, 7 of the 8 candidate genes expressed comprehensive M values below 1.5, with rps18 and ef1a both being ranked first. NormFinder ranks genes based on an overall stability value (SV) and a SV below 1 is considered acceptable. With the exception of ubiquitin (ubiq), all genes produced a SV less than 1, with arginine kinase (ak) being ranked first. BestKeeper ranks genes based on standard deviation (SD); genes with a SD above 1 are considered less stable. Under dsRNA treatment only 4 of the 8 genes produced a SD below one and rps18 was ranked first. Using the delta-Ct method, where lower stability values are considered favorable, ribosomal protein L32 (rpl32) was ranked first. Using the geomean value to create a comprehensive ranking of all four algorithms, with lower values considered more stable, rps18 was ranked first. Given that rps18 was ranked first using two of the four algorithms as well as comprehensively (RefFinder), it was deemed the most stable and selected as a reference gene for our study. A second reference gene, ef1a, was also chosen based on its rankings under GeNorm (first) and Bestkeeper (second), as well as its comprehensive ranking of second. rps18 and ef1a produced corroborating results during gene expression studies. Although our study focused on dsRNA treated beetles for reference gene selection, these results identify potential genes for other gene expression studies using light, temperature, and sex as parameters. rps18 ranked highly under all treatments and has a comprehensive treatment ranking of 1. rpl32 ranks highly under light treatments, while rpl13 does well under temperature treatments, and ak ranks highly when comparing male and female gene expression (for treatment-specific rankings for each method, metadata summary, and raw Cq values, see Supplemental Materials).
Analysis of target gene expression
Of the three target genes evaluated, heat shock protein (hsp), shibire (shi), and inhibitor of apoptosis (iap), hsp and iap have the greatest relative expression and shi has the lowest (Fig. 2). There is no detectable difference in expression between hsp and iap, but both differ significantly from shi. Higher gene expression levels do not necessarily make hsp and/or iap better RNAi targets for inducing mortality. Highly expressed genes may produce shorter half-life proteins and thus require a higher level of expression to perform necessary functions, making them more susceptible to RNAi24, whereas genes with lower levels may produce longer half-life proteins, and therefore do not need to be highly expressed.

Relative expression of hsp, shi, and iap in adult SPB. Relative expression of hsp and iap is higher than that of shi (means followed by the same letter do not differ, one way ANOVA, F2,17 = 36.7, Tukey’s P < 0.0001).
Beetle mortality
With the exception of a single beetle lost in the dsHSP treatment, which may be attributed to experimental handling, no mortality was observed in any dsRNA treatment after 24 hours. After 5 days both dsHSP and dsSHI treated beetles exhibited 40% mortality, which rose to 80% and 73.33% respectively on day 8. At 10 days dsHSP treated beetles experienced 100% mortality, and dsSHI beetles experienced 86.67% mortality (Fig. 3). The difference in mortality between hsp and shi was not significant (t-test, one tailed P = 0.65), but both mortalities were significant relative to control beetles (t-test, one tailed, P = 0.0005). Mortality in dsIAP treated beetles did not differ from control beetles (t-test, one tailed P = 0.72).

Adult SPB survival 10 days after ingesting 1 µL of 10 µL of dsRNA. At day 5, significant differences in survival were evident for beetles ingesting dsHSP and dsSHI relative to those ingesting dsIAP and dsGFP (ANOVA, F3,39 = 4.69, P = 0.0073).
Gene silencing
To assess whether beetle mortality was caused by an RNAi response, gene expression analysis was performed 24 h after dsRNA ingestion using both rps18 and ef1a as internal standards, which showed corroborating results. Ingestion of dsHSP and dsSHI by adult beetles resulted in significant silencing of nearly 50% when compared to expression in control beetles (Fig. 4), whereas ingestion of dsIAP did not result in significant silencing of the iap gene. Our gene silencing results corroborate the mortality produced in hsp and shi treated beetles. The lack of iap silencing and the inability to induce mortality in iap treated beetles further supports that the mortality we observed was a result of gene silencing using the RNAi pathway. However, our iap mortality and gene silencing results differ from studies with emerald ash borer (EAB), another forest pest in which iap is a highly effective target gene16; higher concentrations, longer exposure of dsIAP, or the use of additional dsIAP fragment(s) may produce results similar to those found in EAB. The lack of significant mortality in dsIAP treated beetles corroborates results from our gene expression studies, and demonstrates that iap is not a suitable target gene for SPB under our experimental parameters.

Ingestion of gene specific dsRNAs by adult SPB fed with 1 µL of 10 µg/µL of dsRNA, or dsGFP as a control, resulted in significant silencing of A) hsp (P = 0.0445) and B) shi (P = 0.0013) (one-tailed t-test), but not C) iap (P = 0.3033) (* denotes significant differences).
In conclusion, ours is the first study to demonstrate an RNAi response in southern pine beetle, which led to 100% beetle mortality after ingestion of dsRNA. Although gene silencing in agricultural pests has been studied extensively25,26,27,28, an understanding of the pervasiveness and efficacy of RNAi mechanisms in forest pests is lacking. Initial work with SPB adults demonstrates that a single oral delivery of dsSHI and dsHSP, chosen based on precedence with other forest pests16,29 results in 86.6% and 100% mortality respectively after 10 days. These results align with studies using emerald ash borer neonates29 and red flour beetle (Tribolium castaneum)30. However, our study differs from that investigating EAB in that EAB neonate feeding lasted 4 consecutive days and generated 80% and 93.3% mortality. And though the T. castaneum study generated mortality of 100%, the beetles were injected rather than fed. Oral delivery of dsRNA is less invasive and largely reduces mechanical injury to the insect, but can be less reliable as gut morphology may prevent the delivery of the dsRNA to the midgut epithelium31 and dsRNA degrading enzymes can be present in the gut32. Our results suggest that silencing of the target genes hsp and shi is efficient in SPB, and that SPB may be particularly sensitive to RNAi. Future efforts evaluating RNAi in SPB should focus on screening additional candidate target genes to ensure selection of the most rapid and efficacious for SPB suppression. We recognize that not all effective target genes need lead to high mortality. For example, targeting genes for reproductive suppression or genes leading to a gradual decline in fitness may also prove suitable. Optimal target gene selection is essential for development of effective RNAi technologies to combat southern pine beetle, whose impacts are exacerbated by land management practices33, and changing temperature and precipitation regimes34. Dose responses and combinations of target genes should also be investigated, as these factors have played a role in RNAi efficacy in other forest pests29. Development and deployment of innovative gene silencing technologies such as RNAi in combating forest pests could provide a much-needed tool for natural resource managers, providing an additional component for integrated pest management while minimizing off target effects. Our study is the first step in investigating the feasibility of the use of RNAi against southern pine beetle.
Methods and Materials
Insects
Adult beetles were obtained from loblolly pine, Pinus taeda, bark samples collected from areas with high SPB populations in the southeast USA and stored in darkness at 4 °C. Infested bark was removed as needed, and placed in emergence containers consisting of a sealed, darkened 2 L container with a clear 25 ml collection tube in one end containing a moistened tissue. Emergence containers were held at 23 °C and monitored daily, and only newly emerged adult beetles were used in bioassays.
Reference gene(s)
Candidate gene treatments and selection
Eight reference genes were selected as candidate reference genes: ribosomal protein S18 (rps18), elongation factor – 1 alpha (ef1a), ribosomal protein L13 (rpl13), arginine kinase (ak), succinate dehydrogenase flavoprotein (sdf), ribosomal protein L32 (rpl132), tubulin (tub), and ubiquitin (ubiq)16,35,36. Gene expression was analyzed in individual adult beetles (N = 5) while controlling for four parameters: temperature, light, sex, and exposure to dsRNA, or RNAi response. Temperature treatment beetles were kept at 20 °C and 25 °C. Light treatment beetles were maintained in either total darkness or at 15:9 (L:D). Beetles maintained in both temperature and light regimes were kept in layers of bark and tissue in standard size petri dishes (60 × 15 mm) and were evaluated after three days. Male and female beetles were sexed upon emergence using the presence of pronotal grooves (female) and frontal tubercles (male)37 and used in experiments immediately after sexing. For RNAi exposure, individual adult beetles (N = 5) were fed four separate dsRNA treatments (hsp, shi, iap, and gfp) of 10 µg/µL in a 1% sucrose solution. Following dsRNA ingestion, beetles from each treatment were placed together in petri dishes containing damp filter paper and pine bark; dishes were oriented vertically and maintained at 23 °C with a 15:9 L:D photoperiod. Beetles were evaluated after 24 hours.
Stability analysis
Stability of candidate genes was evaluated by inputting the mean Cq (quantification cycle) value of each beetle per primer into BestKeeper38, an Excel based tool which uses pair-wise comparisons to evaluate gene stability, and into the web based tool, RefFinder39, which integrates four separate algorithms (Table 1) to determine the stability: GeNorm, NormFinder, BestKeeper, and the delta-Ct method. GeNorm measures stability by taking the geometric average and mean pairwise variation of all candidate genes, the results of which produce an M-score (M). Lower M-scores denote higher stability; and genes with scores greater than 1.5 are not considered40. GeNorm selects the best pair of genes, rather than the best single gene. NormFinder produces an overall stability value (SV) by measuring intra and intergroup variations of candidate genes. As with GeNorm, the lower the value, the more stable the gene, with 1 being the cutoff41. BestKeeper measures the standard deviation (SD) of each gene. Again, lower scores denote higher stability, and genes with a SD greater than 1 are considered less stable42. The delta-Ct method is a comparative method that estimates stability based on delta-Ct value variation. Again, lower scores denote greater stability43. RefFinder calculates a comprehensive final ranking of the geometric mean of the four algorithms with smaller geometric means denoting higher stability44.
RNA extraction, cDNA synthesis, and qPCR
Total RNA was isolated from whole beetles with TRI Reagent RT (Molecular Research Center Inc., Cincinnati, OH), RNA integrity was verified using gel electrophoresis and absorbance was measure at 260/280 and 230/280. cDNA was synthesized using SuperScript¨ III Reverse Transcriptase (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions at a concentration of 3000 ng/ml and used as a template for the qPCR standard curve, constructed using a 5-fold dilution. Each qPCR sample contained 1 µL of 3000 ng/µL synthesized cDNA (diluted 1:1), 0.2 µL of each primer (forward and backward), 3.6 µL of nuclease free ddH2O, and 5 µL of SYBR Green PCR Master Mix (Applied Biosystems, USA); totaling 10 µL. All reactions were performed using SYBR Green Master Mix and amplified under the following cycling conditions: beginning cycle at 95 °C, 40 cycles at 95 °C for denaturation, followed with 30 s at 65 °C for annealing and extension, and ending with generation of a melting curve consisting of a single peak to rule out non-specific product and primer dimer formations. Each sample was repeated three times and measured using the mean Cq value. For evaluating reference genes, the mean Cq value of each sample and each primer was used as input data. For gene expression analysis the 2−∆∆Ct method45 was used to calculate the relative expression level of the target gene with rps18 and ef1a as reference genes. For statistical analysis, we performed a one-way ANOVA with Tukey’s HSD to evaluate differences (P < 0.05). Primers for the eight selected genes (Table 2) as well as three target genes (Table 3) were designed using Primer3Plus and validated using correlation coefficients (R2) and amplification efficiencies (Eff). Standard curves were constructed using 5-fold serially diluted cDNA for each pair of primers. A desired R2 is >0.99 and acceptable amplification efficiencies fall between 90% and 110%46.
dsRNA synthesis from cDNA
Gene specific primers (Table 3) designed using SnapDragon were used to amplify dsRNA templates, and run under normal PCR conditions as follows: 4 min at 94 °C, followed by 35 cycles of 30 sec at 94 °C, 30 sec at 60 °C, and 45 sec at 72 °C. The final step is an extension incubation which takes place at 72 °C for 10 min. PCR templates were purified using a Qiagen purification kit (Qiagen, Germantown, MD). Once purification was completed, MEGAscript RNAi Kit (Thermo Scientific, Waltham, MA) was used in dsRNA synthesis, per manufacturer’s instructions. The reaction mix was then incubated at 37 °C for 14 hours; after which it underwent 30 minutes of DNase treatment at 37 °C. dsRNA was recovered by adding 2 µL (0.1 × volume) of sodium acetate and 50 µL (2.4 × volume) of 100% EtOH to the reaction mix which was then incubated at −20 °C for 2 hours. After incubation, the mix was spun at −4 °C for 30 min (14000 rpm), then washed with 750 µL of 75% EtOH and spun at −4 °C for 15 min at 13000 rpm. Once rinsed, samples were dried at 37 °C for 25 min and re-suspended in 20 µL of nuclease free H2O. dsRNA quality was checked using electrophoresis and quantified with a spectrophotometer (NanoDrop Technologies, Wilmington, DE). To attain a desired concentration of 10 µg/µL, dsRNA was dried using vacuum speed at 30 °C for 15 minutes and resuspended in nuclease free H2O.
Adult beetle feeding assays
To evaluate adult SPB mortality and gene silencing, adult beetles (N = 15 per treatment for mortality assay; N = 5 per treatment for gene silencing) were fed dsRNA diluted in sucrose. A 1 µL drop of 10 µg of dsRNA in a 1% sucrose solution colored with blue food coloring for easier visualization was placed on the wall of a 50 mm petri dish (Fig. 5)15. Individual beetles were then placed in the petri dish so that their mouthparts were in contact with the droplet and held in place with a paintbrush, applying slight pressure to the back of the head. Each beetle was held in place until the solution was consumed (3–5 min). Beetles were fed one at a time to ensure complete ingestion of the droplet.

Adult SPB feeding on sucrose droplet.
Once dsRNA ingestion was complete, beetles from each treatment were placed together in petri dishes containing damp filter paper and pine bark; dishes were then oriented vertically beneath a clear plastic chamber and maintained at 23 °C with a 15:9 L:D photoperiod. Beetles were monitored for 10 days, with mortality being recorded every 24 hours. For gene silencing analysis, beetles were collected after the first 24 hours, at which time total RNA was extracted. For statistical analysis of beetle mortality, a one way ANOVA on non-transformed data was performed, and Tukey’s test was used to identify differences between treatments. For gene silencing analysis, data were determined to be normal (Shapiro-Wilk test, W = 0.6652), and a one-tailed t-test was used to compare differences of a single variable.
References
Holmes, T. P. Price and welfare effects of catastrophic forest damage from southern pine beetle epidemics. Forest Sci. 37, 500–516 (1991).
Pye, J., Holmes, T., Prestemon, J. & Wear, D. Economic impacts of the southern pine beetle. In Coulson, R.N. & Klepzig K.D. Southern Pine Beetle II. U.S. Department of Agriculture Forest Service, Southern Research Station, Gen. Techn. Rep. SRS-140, Asheville, NC. Pp 213–222 (2011).
Leuscher, W. A. & Young, R. L. Estimating the southern pine beetle’s impact on reservoir campsites. Forest Sci. 24, 527–537 (1978).
Schowalter, T. D., Coulson, R. N. & Crossley, D. A. Role of the southern pine beetle and fire in maintenance of structure and function of the southeastern coniferous forest. Environ. Entomol. 10, 821–825 (1981).
Drooz, A. T. Insects of eastern forests. U.S. Department of Agriculture Forest Service Misc. Publ. 1426, Washington DC (1985).
Pacala, S. W., Canham, C. D., Silander, J. A. & Kobe, R. K. Sapling growth as a function of resources in a northern temperate forest. Can. J. For. Res. 24, 2172–2183 (1984).
Palik, B. J., Mitchell, R. J., Housead, G. & Pederson, N. Effects of the canopy structure on resource availability and seedling responses in a longleaf pine ecosystem. Can. J. For. Res. 27, 1458–1464 (1997).
McGuire, J. P. et al. Gaps in a gappy forest: Plant resources, longleaf pine regeneration, and understory response to tree removal in longleaf pine savannas. Can. J. For. Res. 31, 765–778 (2001).
Coleman, T., Clarke, S., Meeker, J. & Rieske, L. K. Forest composition following overstory mortality from southern pine beetle and associated treatments. Can. J. For. Res. 38, 1406–1418 (2008).
Tchakerian, M. D. & Coulson, R. N. Ecological impacts of southern pine beetle. In Coulson, R.N. & Klepzig K.D. Southern Pine Beetle II. U.S. Department of Agriculture Forest Service, Southern Research Station, Gen. Techn. Rep. SRS-140, Asheville, NC. Pp. 223–234 (2011).
Liebhold, A. & Williams, D. Climate change and the outbreak ranges of two North American bark beetles. Agric. For. Entomol. 4, 87–99 (2002).
Lesk, C., Coffel, E., D’Amato, A. D., Dodds, K. & Horton, R. Threats to North American forests from southern pine beetle with warming winters. Nat. Clim. Change 7, 713 (2017).
Dietrich, I. et al. The antiviral RNAi response in vector and non-vector cells against orthobunyaviruses. PLOS Neglected Tropical Diseases 1, 5, https://doi.org/10.1371/journal. pntd.ooo5272 (2017).
Zhao, C., Gonzales, M. A. A., Poland, T. M. & Mittapalli, O. Core RNAi machinery and gene knockdown in the emerald ash borer (Agrilus planipennis). J. Insect Physiol. 72, 70–78 (2015).
Rodrigues, T. B., Rieske, L. K., Duan, J. J., Mogilicherla, K. & Palli, S. R. Development of RNAi method for screening candidate genes to control emerald ash borer, Agrilus planipennis. Sci. Rep. 7, https://doi.org/10.1038/s41598-017-07605-x (2017).
Rodrigues, T. B., Dhandapani, R. K., Duan, J. J. & Palli, S. R. RNA interference in the Asian longhorned beetle: Identification of key RNAi genes and reference genes for RT-qPCR. Sci. Rep. 7, https://doi.org/10.1038/s41598-017-08813-1 (2017).
Whyard, S., Singh, A. D. & Wong, S. Ingested double-stranded RNAs can act as species-specific insecticides. Insect Biochem. Mol. Biol. 39, 824–832.
Zhu, F., Xu, J., Palli, R., Ferguson, J. & Palli, S. R. Ingested RNA interference for managing the populations of the Colorado potato beetle, Leptinotarsa decemlineata. Pest Manage. Sci. 67, 175–182 (2011).
Palli, S. R. RNA interference in Colorado potato beetle: steps toward development of dsRNA as a commercial insecticide. Curr. Opin. Insect Sci. 6, 1–8 (2014).
Huvenne, H. & Smagghe, G. Mechanisms of dsRNA uptake in insects and potential of RNAi for pest control: A review. J. Insect Physiol. 56, 227–235 (2010).
Yu, N. et al. Delivery of dsRNA for RNAi in insects: An overview and future directions. Insect Sci. 20, 4–14 (2013).
Baum, J. A. et al. Control of coleopteran insect pests through RNA interference. Nat. Biotech 25, 1322, Nbt1359#supplementary-information, https://www.nature.com/articles (2007).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geo- metric averaging of multiple internal control genes. Genome Biol. 3, 10–1186, https://doi.org/10.1186/gb-2002-3-7-research0034 (2002).
Hong, S. W., Jiang, Y., Kim, S., Li, C. J. & Lee, D. K. Target gene abundance contributes to the efficiency of siRNA-mediated gene silencing. Nucleic Acid Therapeutics 24, 192–198, https://doi.org/10.1089/nat.2013.0466 (2014).
Bolognesi, R. et al. Characterizing the mechanism of action of double-stranded RNA activity against western corn rootworm (Diabrotica virgifera virgifera LeConte). PLoS One 7, e47534, https://doi.org/10.1371/journal.pone.0047534 (2012).
Palli, S. R. RNAi methods for management of insects and their pathogens. DNA 5, 3 (2012).
Galdeano, D. M., Breton, M. C., Lopes, J. R., Falk, B. W. & Machado, M. A. Oral delivery of double-stranded RNAs induces mortality in nymphs and adults of Asian citrus psyllid, Diaphorina citri. Plos One 12, 0171847, https://doi.org/10.1371/journal.pone.01 (2017).
Miller, S. C., Miyata, K., Brown, S. J. & Tomoyasu, Y. Dissecting systemic RNA interference in the red flour beetle Tribolium castaneum: Parameters affecting efficiency of RNAi. Plos One 7, https://doi.org/10.1371/journal.pone.0047431 (2012).
Rodrigues, T. B., Duan, J. J., Palli, S. R. & Rieske, L. K. Identification of highly effective target genes for RNAi-mediated control of emerald ash borer, Agrilus planipennis. Sci. Rep. 8, 520, https://doi.org/10.1038/s41598-018-23216-6 (2018).
Ulrich, J. et al. Large scale RNAi screen in Tribolium reveals novel target genes for pest control and the proteasome as prime target. BMC Genomics 16, 674, https://doi.org/10.1186/s12864-015-1880-y (2015).
Scott, J. G. et al. Towards the elements of successful insect RNAi. J. Insect Phys. 59, 1212–1221, https://doi.org/10.1016/j.jinsphys.2013.08.014 (2013).
Arimatsu, E., Kotani, Y., Sugimura, Y. & Furusawa, T. Molecular characterization of a cDNA encoding dsRNase and its expression in the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 37, 176–183, https://doi.org/10.1016/j.ibmb.2006.11.004 (2007).
Nowak, J. T., Meeker, J. R., Coyle, D. R., Steiner, C. A. & Brownie, C. Southern pine beetle infestation in relation to forest stand condition, previous thinning, and prescribed burning: evaluation of the southern pine beetle prevention program. J. For. 113, 454–462 (2015).
Weed, A., Ayres, M. & Hicke, J. Consequences of climate change for biotic disturbances in North American forests. Ecol. Monograph 83, 441–470 (2013).
Rodrigues, T. B. et al. Validation of reference housekeeping genes for gene expression studies in western corn rootworm (Diabrotica virgifera virgifera). PLoS ONE 9, 109825, https://doi.org/10.1371/journal.pone.0109825 (2014).
Koramutla, M. K., Aminedi, R. & Bhattacharya, R. Comprehensive evaluation of candidate reference genes for qrt-pcr studies of gene expression in mustard aphid, Lipaphis erysimi (Kalt). Sci. Rep. 6, https://doi.org/10.1038/srep25883 (2016).
Osgood, E. A. & Clark, E. W. Methods of sexing and sex ratios of the southern pine beetle. Dendroctonus frontalis Zimm. Can. Entomol. 95, 6–10 (1963).
Pfaffle, M., Tichopad, A., Prgomet, C. & Neuvians, T. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – Excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 47, https://doi.org/10.1023/B:BILE.0000019559.84305 (2004).
Spiegelaere, W. D. et al. Reference gene validation for RT-qPCR, a note on different available software packages. PloS One 10, https://doi.org/10.1371/journal.pone (2015)
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 18, https://doi.org/10.1186/gb-2002-3-7-research0034 (2002).
Anderson, C. L., Jensen, J. L. & Orntoft, T. F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify if genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res., https://doi.org/10.1158/0008-5472.CAN-04-0496 (2004).
Pfaffl, M. W., Tichopad, A., Prgomet, C. & Neuvians, T. P. Determination of stable housekeep- ing genes, differentially regulated target genes and sample integrity: Bestkeeper – excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–515 (2004).
Silver, N., Best, S., Jing, J. & Thein, S. L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 7, 33 (2006).
Spiegelaere, W. D. et al. Reference gene validation for RT-qPCR, a note on different available software packages. PloS ONE, https://doi.org/10.1371/journal.pone.0122515 (2015).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(t)(-delta delta c) method. Methods 25, 402–408 (2001).
Broeders, S. et al. Guidelines for validation of qualitative real-time PCR methods. Trends in. Food Sci. Technol. 37, 115–126, https://doi.org/10.1016/j.tifs.2014.03.008 (2014).
Acknowledgements
The authors thank David Coyle, Wood Johnson, and William Shepherd (USDA Forest Service) for assistance obtaining beetles, and Bruce Webb (University of Kentucky) for comments on an early version of this manuscript. This is publication number 18-08-070 from the Kentucky Agricultural Experimental Station and is published with the approval of the director. This work was supported by the University of Kentucky, the USDA Forest Service Forest Health Research and Education Center, and the Kentucky Agricultural Experiment Station under McIntire-Stennis 2351197000.
Author information
Authors and Affiliations
Contributions
B.R.K. conducted the experiments. B.R.K., T.B.R., and L.K.R. conceived the experiments, analyzed and interpreted the results, and prepared the manuscript. All authors approve of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kyre, B.R., Rodrigues, T.B. & Rieske, L.K. RNA interference and validation of reference genes for gene expression analyses using qPCR in southern pine beetle, Dendroctonus frontalis. Sci Rep 9, 5640 (2019). https://doi.org/10.1038/s41598-019-42072-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42072-6
This article is cited by
-
Double-stranded RNA (dsRNA) technology to control forest insect pests and fungal pathogens: challenges and opportunities
Functional & Integrative Genomics (2023)
-
Southern pine beetle-specific RNA interference exhibits no effect on model nontarget insects
Journal of Pest Science (2022)
-
Validation of reference genes for quantitative PCR in the forest pest, Ips calligraphus
Scientific Reports (2021)
-
Recent advances toward the sustainable management of invasive Xylosandrus ambrosia beetles
Journal of Pest Science (2021)
-
Confocal microscopy provides visual evidence and confirms the feasibility of dsRNA delivery to emerald ash borer through plant tissues
Journal of Pest Science (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
