
Abstract
Obesity is a high risk factor for colorectal cancer (CRC). The contribution of underlying epigenetic mechanisms to CRC and the precise targets of epigenetic alterations during cancer development are largely unknown. Several types of epigenetic processes have been described, including DNA methylation, histone modification, and microRNA expression. To investigate the relationship between obesity and CRC, we studied both obese and CRC patients, focusing on genome-wide peripheral blood DNA methylation alterations. Our results show abnormal distributions of overlapping differentially methylated regions (DMRs) such as hypermethylated CpG islands, which may account for epigenetic instability driving cancer initiation in obesity patients. Furthermore, functional analysis suggests that altered DNA methylation of extracellular (e.g., O-glycan processing) and intracellular components contribute to activation of oncogenes (e.g. KRAS and SCL2A1) and suppression of tumor suppressors (e.g. ARHGEF4, EPHB2 and SOCS3), leading to increased oncogenic potency. Our study demonstrates how DNA methylation changes in obesity contribute to CRC development, providing direct evidence of an association between obesity and CRC. It also reveals the diagnostic potential of using DNA methylation as an early risk evaluation to detect patients with high risk for CRC.
Similar content being viewed by others

Introduction
Being overweight or obese is considered to be a major risk factor for many cancers, in particular colorectal cancer (CRC)1,2,3. Epidemiological data suggests that obesity is associated with a 1.2–2.0 fold increased risk of CRC4. Even though the close link between obesity and the risk of CRC has been suggested by a large number of studies5,6,7,8,9, the underlying molecular mechanisms are still largely unknown. Understanding the mechanisms linking obesity to the development of CRC may lead to the development of accurate methods for early detection and the identification of new targets for CRC prevention.
DNA methylation is an epigenetic mechanism that occurs when a methyl group is added onto the C5 position of cytosine, thereby modifying gene function and affecting gene expression10,11,12. Most DNA methylation occurs at cytosine residues that precede guanine residues, called CpG dinucleotides, which tend to cluster in DNA domains known as CpG islands. The relationship between methylation and gene expression is complex. In general, DNA methylation of gene promoters is associated with transcriptional silencing13, whereas methylation in gene bodies is associated with increased gene expression14,15,16. Strong correlations between gene expression and CpG islands and island shores have been demonstrated17. Inappropriate methylation of CpG islands could result in impaired transcription factor binding, recruiting repressive methyl-binding proteins, and stably silencing gene expression10. Global hypomethylation is thought to influence CRC development by inducing chromosomal instability18,19,20.
Compared to studies in cancer, studies in obesity have not provided consistent evidence of a role for global methylation changes. Furthermore, differentiating early epigenetic alterations potentially involved in cancer initiation is difficult considering the influence of multiple other factors on these epigenetic changes. Consequently, studying specific methylation changes that affect oncogenic transformation signaling is likely to provide a better picture of the association between obesity and CRC development.
Genome-wide mapping of differentially methylated CpG sites (DMCs) or differentially methylated regions (DMRs) is an important means to reveal the impact of epigenetic modifications on inheritable phenotypic variation in both obesity and CRC and to understand their correlation. Currently, a massive effort is directed at providing better insight into tissue-specific epigenetic alternations and their roles in disease development21,22,23,24,25. Ronn, T. et al. demonstrated that epigenetic biomarkers in blood can mirror epigenetic signatures in target tissues21. Using bisulfite pyrosequencing, Ally and colleagues observed a correlation between colonic tissue methylation and blood methylation of estrogen receptor 1 (ESR1) that is independent of age, gender, disease status, and body mass index (BMI)26. To date, only a few studies have reported results from examining the genome-wide methylation pattern in colorectal tumors27,28,29,30 and no earlier studies have specifically addressed the effects of DNA methylation alterations in the blood of CRC patients. The aim of the present study was to explore whole blood DNA methylation patterns in obese and CRC patients to identify epigenetic changes associating CRC to obesity by comparing whole genomic DMR and DMC patterns of DNA methylation using an overlapping method. We provide direct evidence of the connection between cancer development and obesity. The recognition that the same epigenetic changes are a driving force for the development into CRC in obese individuals supports the promising biomarker potential of DNA methylation studies for early diagnosis.
Results
Significant associations observed between obesity and CRC in Overlapping DMCs and DMRs
Genome-wide methylation analysis was conducted in 15 CRC patients and compared to publically available data from 10 obese subjects and 15 healthy lean controls (Table 1). The case and control groups were comparable with respect to gender. Age was used as a covariate in differential analysis in order to remove its possible effects. To avoid systematic errors for the DNA methylation data, histogram transformation was applied to equalize the distributions of the methylation levels to the control group. We performed differential methylation analysis of the reduced representation bisulfite sequencing (RRBS) profiling data for the obesity and CRC cases versus the control group. We identified 186,511 DMCs between CRC and control subjects and 91,809 DMCs between obese and controls (Fig. 1). To evaluate whether obesity is associated with CRC through DNA methylation alterations, we overlapped these DMCs identified separately from CRC and obesity. If CRC and obese DNAs were both differentially methylated at a certain CpG site, the DMC was counted as an overlapping DMC. As shown in Fig. 1A, there were 40,605 overlapping DMCs, accounting for 44% of DMCs identified from the obese group. Surprisingly, we observed methylation changes of these overlapping DMCs occurred in the same direction for obesity and CRC (Fig. 1B). In other words, a vast majority of overlapping DMCs for obesity and CRC were either hypermethylated (36.7%) or hypomethylated (45.2%) for both obesity and CRC (Fig. 1C). A Chi-square test showed that there was a significant association between obesity and CRC (p < 0.0001).

Association of differentially methylated CpGs (DMCs) in obesity and CRC. (A) Venn-diagram of DMCs generated from CRC vs. Control and Obesity vs. Control genes; (B) Scatter plot displaying the methylation differences in overlapping DMCs and the distribution of these DMCs partitioned by hyper-/hypomethylated CpGs in CRC and obesity; (C) Number of hyper-/hypomethylated CpGs in CRC and obesity. The Chi-Square test was used to determine a potential significant relationship between obesity and CRC.
A similar trend of DNA methylation alterations was observed in the 750 overlapping DMRs identified (Fig. 2). They accounted for 27.6% of the 2,713 DMRs identified in the obese group. The association between obese and CRC in overlapping DMRs was highly significant as suggested by Chi square test (p < 0.0001). These data suggest that obesity is highly associated with the risk of developing CRC.

Association of differentially methylated regions (DMRs) in obesity and CRC. (A) Venn-diagram of the DMRs generated from CRC vs. Control and Obesity vs. Control genes; (B) Scatter plot displaying the mean methylation difference of overlapping DMRs and the distribution of these DMRs partitioned by hyper-/hypomethylated DMRs in CRC and obesity. (C) Number of hyper-/hypomethylated DMRs in CRC and obesity.
Distribution patterns in overlapping DMRs is Similar to the pattern in CRC
We then annotated hypermethylated or hypomethylated overlapping DMRs for both obesity and CRC to gene regions and CpG islands (Fig. 3). In CRC, peripheral blood showed higher overall genomic hypomethylation than hypermethylation21,22,23,24 (Fig. 3A). This is consistent with previous reports showing that genomic DNA hypomethylation is a hallmark of most cancer genomes, prompting genomic instability and cancer transformation20,25,26,27,28. In contrast, the numbers of hyper- or hypomethylated DMRs in all gene regions were similar in obesity (Fig. 3B). However, the distribution of overlapping DMRs (Fig. 3C) was closer to the distribution of non-overlapping DMRs in cancer than the distribution of non-overlapping DMRs in obesity. Greater number of hypomethylated rather than hypermethylated DMRs were identified in the promoter, intron and intergenic regions in cancer and overlapping DMRs (Fig. 3A,C). Similarly, a closer distribution was seen among the distribution of overlapping DMRs and the distribution of non-overlapping DMRs in cancer over CpG islands (Fig. 3D–F).

Bar plots of the number of cancer-specific, obesity-specific, overlapping DMRs by gene subregions (A–C) and CpG islands (D–F).
These data suggest that the risk of obesity-related CRC can be potentially be evaluated by analyzing overlapping DMRs. Using gene annotation enrichment analysis, biomarkers linking obesity to CRC risk can be potentially identified.
Functional analysis of genes associated with overlapping DMRs
To identify the most important DMRs linking obesity to CRC, DMRs either hyper- or hypomethylated for both obesity and CRC were further selected using nonparametric procedures. The average methylation level across DMRs was calculated for each subject, and the Kruskal-Wallis test, followed by multiple pair-wise comparisons of groups by Mann-Whitney U test, was performed to determine significance of differences. Four hundred and forty-two DMRs with a p value of at least 0.1 or less by the Kruskal-Wallis and Mann-Whitney U tests were selected. Among these, 238 DMRs were located in the promoter or gene body regions.
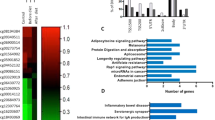
KEGG pathway29,30,31 and GO biological process (GO-BP) analyses of the 238 genes associated with overlapping DMRs were performed to better understand how DNA methylation links obesity to CRC development. The top KEGG pathways and GO-BP are summarized in Table 2. Overall, these analyses, enriched by selected genes, are related to the extracellular microenvironment such as the extracellular matrix microbiota and mucin glycans, and to changes in intracellular signaling pathways, such as metabolic, transforming growth factor (TGF)-β and KRAS signaling, which may play a central role in CRC initiation32,33.
Major pathways affected by DNA methylation
The signal-transduction pathways dysregulated by DNA methylation changes in both obesity and CRC include: 1) extracellular matrix components, i.e., O-glycan processing, protein glycosylation, and extracellular matrix scaffold; 2) KRAS and TGF-β signaling; and 3) lipid and glucose metabolism (Table 2 and Fig. 4). The alterations in these extracellular and intracellular metabolites could induce CRC-associated metabolic reprogramming in obesity and contribute to the initiation of CRC in obese patients. Further details are given below.

Mean methylation levels of relevant DMRs. (A) Summary of 12 overlapping major DMRs; (B–M) Box plots of methylation level of individual DMRs in three different gene groups. Each dot represents the mean methylation level of the specific DMR for each individual subject.
Extracellular matrix components affected by methylation changes
In the O-glycan processing (GO: 0016266) and mucin type O-glycan biosynthesis pathway (hsa00512), 5 genes were associated with selected DMRs. β-1,4-galactosyltransferase 5 (B4GALT5) (intron region), polypeptide N-Acetyl galactosaminyl transferase 17 (GALNT17) (intron region) and mucin 5B (MUC5B) (coding region) (Fig. 4B). These genes were hypomethylated, whereas glucosaminyl (N-acetyl) transferase 1 (GCNT1), GALNT6 were hypermethylated in the 5′-UTR and promoter regions, respectively. The CRC microenvironment and extracellular matrix are mainly constituted by collagen and elastin34,35,36. In the extracellular matrix organization (GO: 0030198), we observed that the elastin (ELN), and collagen α1 (IX) chain (COL9A1) (Fig. 4C) in the coding region were hypomethylated, and COL6A1 was hypermethylated in the coding region. Elastin and collagen are the main components of elastic fibers and their DNA methylation changes indicate altered extracellular matrix barrier function which may be associated with tumor progression37,38.
Altered DNA methylation of KRAS and TGF-β signaling
In the pathways in cancer (hsa05200), although not in the list of top KEGG pathways, we found 10 genes with selected DMRs, among which 4 genes are involved in the CRC pathway (hsa05210) (see Table 2). These include TGF-β2, KRAS (Fig. 4D), adenomatous polyposis coli protein 2 (APC2) and SMAD family member 3 (SMAD3) (Fig. 4E). KRAS, a well-established proto-oncogene, was hypermethylated in the intron region. We also found that fibroblast growth factor 3 (FGF3) (Fig. 4F), upstream of KRAS, is associated with a hypomethylated DMR in its promoter region in obesity and is even further hypomethylated in CRC. SMAD3, a central component of the TGF-β signaling pathway39, was hypomethylated in the promoter region. Solute carrier family 2 member 1 (SLC2A1), which encodes the glucose transporter type 1 protein (GLUT1) and is responsible for basal glucose transport in all cell types, contained a hypomethylated DMR in its 5′-UTR region (Fig. 4G). APC-stimulated guanine nucleotide-exchange factor (ARHGEF4) was hypermethylated in the promoter region in CRC peripheral blood (Fig. 4H). The epidermal growth factor receptor (EGFR), a transmembrane tyrosine kinase involved in triggering the MAPK signaling pathway40, was associated with a hypomethylated DMR in its coding region (Fig. 4I). Eph receptor B2 (EPHB2) has been suggested to be a tumor suppressor gene in colorectal carcinogenesis41, and was associated with a hypomethylated DMR in its intron region (Fig. 4J).
Aberrant DNA methylation of lipid and glucose metabolism genes
Obesity is related to energy imbalance and metabolic dysfunction42,43. Consistent with this, we observed some related genes associated with overlapping DMRs. Regulatory-associated protein of mTOR (RPTOR) is involved in the control of mTORC1 activity, which plays an important role in lipogenesis and in regulating the endothelial cell proliferation (GO: 0001938) (see Table 2). We identified hypermethylated overlapping DMR of RPTOR located in the gene body region. In addition, we observed a hypomethylated DMR in the coding region of suppressor of cytokine signaling 3 (SOCS3) (Fig. 4K). We also observed methylation changes in several mitochondria-related genes, such as hydroxyacyl-Coenzyme A dehydrogenase (HADH), an enzyme that catalyzes the metabolism of short-and medium-chain fatty acids44. This showed a hypomethylated DMR in the gene body region in obesity which was further hypomethylated in CRC, representing changes that may affect its function in lipid metabolism. The succinate dehydrogenase (SDH) complex (SDHAF1), which encodes a protein essential for the assembly of mitochondrial enzyme succinate dehydrogenase (SDH), the main element of complex II45, was hypermethylated in its promoter region in obesity and further hypermethylated in CRC. In addition, RXRA, a common heterodimeric partner for a number of nuclear receptors46, was hypermethylated in the intron region in obesity and further hypermethylated in CRC. Consistent with a recent study showing that HIGD1A expression is increased during glucose deprivation to modulate cell survival and tumor growth47, we observed a hypomethylated DMR in the promoter region of HIGD1A in CRC (Fig. 4L). Calcium voltage-gated channel subunit alpha1 C (CACNA1C) belongs to the insulin secretion and MAPK signaling pathways and alterations in its expression may have an adverse effect on tissue homeostasis, which may result in tumorigenesis48. We identified a hypomethylated DMR in the intron region (Fig. 4M). Collectively, aberrant lipogenesis and changes in lipid and glucose metabolism are key features of metabolic reprogramming, which may induce aberrant activation of KRAS signaling and a sustained pro-inflammatory environment, leading to cancer initiation.
Altered mRNA expression of representative genes affected by DNA methylation in HCT116 cells
The DNA methyltransferases, DNMT1 and DNMT3B, are essential for maintenance and de novo CpG methylation and disruption of these 2 genes results in more than 95% loss of genomic methylation49. To further confirm the functional impact of DNA methylation alterations on gene expression, we utilized two human colorectal carcinoma cell lines, namely HCT116 wild type and HCT116 DNMT1/DNMT3B double knockout (DKO) cells. As shown in Fig. 5, gene expression of these representative genes were affected by DNA methylation changes with high reproducibility. We observed an opposite effect on the gene expression of a group of genes (KRAS, FGF3, HIGD1A and SLC2A1) as compared with the other group of genes (ARHGEF4, CACNA1C, EGFR, EPHB2, SOCS3, SMAD3, MUC5B and COL9A1). Especially, KRAS showed reduced expression and ARHGEF4 showed elevated expression in HCT116 DKO as a consequence of DNMT inhibition, consistent with the hypermethylated DMRs we observed in the gene body and promoter regions of KRAS and ARHGEF4, respectively in CRC.

Heatmap displays mean row-centered log-CPM (log2-counts per million) values of the representative genes selected in Fig. 4 in two replicas of HCT116 and DKO cell line.
Discussion
CRC remains the second leading cause of cancer-related death in the United States50. The 5-year relative survival rate for early-stage CRC is 90%; for advanced stage IV CRC, the rate drops to about 11%51. But only about 4 out of 10 CRCs are found at the early stage52, partially due to the poor patient acceptance and/or sensitivity of available screening modalities. Blood-based DNA methylation has great potential as an early, accurate, non-invasive biomarker for risk evaluation and early detection to improve the survival rate for CRC patients. Obesity is a complex disorder that contributes to many human diseases53. In this study, we aimed at understanding whether DNA methylation alterations in blood play a role in the association between obesity and CRC. Using genome-wide methylation sequencing data and overlapping analysis, we observed DNA methylation changes in obesity and CRC with significant association (Figs 1 and 2). The distribution pattern of overlapping DMRs, such as hypermethylated CpG islands, was similar as the pattern of DMRs in CRC were comparable to that in obesity (Fig. 3). By further analyzing these overlapping DMRs, we observed DNA methylation changes in extracellular matrix components and organization, O-glycan processing, and intracellular factors including KRAS signaling and lipid and glucose metabolism, all pathways that may enhance the CRC risk in obesity.
In the extracellular components, we showed DNA methylation changes in the mucin type O-glycan biosynthesis pathway (hsa00512) and O-glycan processing (GO: 0016266) (Table 2). Mucins are the main components of mucus and the colonic mucus forms a protective homeostatic barrier against enteric pathogens between the resident microbiota and the underlying epithelial cells54,55,56,57. These DNA methylation alterations would lead to mucus degradation and compromise epithelial barrier function.
In the intracellular signaling domain, our data indicated altered DNA methylation of KRAS and of metabolic reprogramming, which play a crucial role in tumorigenesis. Our data suggests that metabolic stress in obesity contributes to the acquisition of an oncogenic potential. Alterations in DNA methylation may contribute to dysregulation of the insulin signaling pathway, which is associated with activated oncogenes (e.g. KRAS and SCL2A1) and downregulated tumor suppressors (e.g. SOCS3, EPHB2 and ARHGEF4), leading to increased and unregulated cellular proliferation and malignant transformation. The intron region of KRAS was hypermethylated in both CRC and obesity. KRAS signaling is also a shared component in signaling pathways regulating pluripotency of stem cells, microRNAs in cancer and oxytocin signaling pathway (hsa04550, hsa05206, and hsa04921, respectively), suggesting its central role in obesity and cancer pathology. SCL2A1, which encodes the GLUT1 transporter58, was hypomethylated in its promoter region. GLUT1 is primarily undetectable in normal epithelial tissues and benign epithelial tumors, and overexpression of GLUT1 during oncogenesis has been identified in various cancers, and is considered as an important player of active tumor cell glucose uptake and metabolism59,60. In addition, metabolic stress, such as increased glucose uptake induced by SLC2A1 upregulation and glycolysis, is consistent with oncogenic mutations in oncogenes, such as KRAS or BRAF61. In contrast, the coding region of SOCS3 which is associated with obesity-related cancers, was hypomethylated in obesity and CRC. A previous study suggested that methylation silencing of SOCS3 suppresses its response to IL-6 stimulation and increases the propensity to malignant transformation62. ARHGEF4 is a binding partner of adenomatous polyposis coli (APC)63 and an important tumor suppressor gene in the development of CRC64. We found that ARHGEF4 was hypermethylated in the promoter region, which may suppress its expression. Moreover, we observed the transforming growth factor (TGF)-β /SMAD signaling was disrupted by DNA methylation as a shared component in angiogenesis, negative regulation of TGF-β receptor signaling pathway, regulation of TGF-β production and CRC pathway (GO: 0001525, GO: 0030512, GO: 0032909 and hsa05210, respectively). This disruption may contribute to pro-tumorigenic mechanisms of TGF-β signaling. Previous studies have shown TGF-β signaling reduces proliferation, promotes apoptosis and differentiation and acts as a tumor suppressor in premalignant tumor development and as a tumor promoter in advanced tumors65. SMAD3 can form transcription complexes to regulate TGF-β target genes and loss of SMAD3 appears to promote colorectal tumorigenesis66,67. By utilizing RNA-seq data from HCT116 and DKO cell lines, we further verified the functional consequences of DNA methylation changes of selected gene for colorectal tumorigenesis (Fig. 5). Our results, although may not accurately reflect the role of blood DNA methylation alterations during tumorigenesis, suggest the different impacts of DNA methylation changes on the gene expressions of oncogenes (e.g. KRAS and SCL2A1) and tumor suppressor genes (e.g. ARHGEF4, EPHB2 and SOCS3). In order to fully elucidate the impact of DNA methylation alterations on CRC development and progression, further studies on the physiological function analysis of each genes is warranted.
Despite the above novel findings, some limitations in our study should be noted. Clearly, the sample size is relatively small. We took a more conservative approach to the data analysis. For example, we only selected 238 DMRs for functional analysis. Despite the encouraging initial CRC specific results, further work is warranted to validate these findings in a large cohort of patients.
Furthermore, there is a large difference between the mean ages of the CRC group and the obese or control groups. The association between age and the DNA methylation profile is previously reported. As indicated in the first paragraph of the Results section, age was considered as a covariate in the logistic regression model for detecting differential DNA methylation to control any distortion effect. Given the fact that the overlapping DMRs were identified separately from CRC and obesity and the obese group is age-matched to the control group, we can deduce that age has minimal effect on the overlapping DMRs. Further studies with age-matched CRC patients are needed to determine the relationship between age-related methylation changes and CRC susceptibility.
Finally, we acknowledge the heterogeneity of our sample as whole blood samples contain a mixture of various cells that exist in the blood circulation. Nonetheless, an interplay between cell types composing the whole blood exists and may have an implication for CRC development. It was thus important to assess whole blood rather than isolated plasma, serum and leukocytes including monocytes, macrophages and neutrophils. Moreover, the contribution of the cellular composition is accounted for by the total variation of DNA methylation measured. In clinical research, whole blood is one of the most readily available samples for biomarker analysis. Because under certain circumstances the amount of blood drawn from patients does not allow us to analyze the contribution of each component to DNA methylation changes in blood, a pooling method using DNA from groups of individuals has recently shown promise in identifying significant methylation markers68.
In summary, our study points to DNA methylation alterations linking obesity and CRC with the promise for early prognosis of CRC risk in relation to obesity. Our results provide additional information for deeper understanding of CRC development, and highlight potential new targets for prevention of CRC. Future research effort should include the integration of DNA methylation, gene expression and disease initiation and progression to provide comprehensive insight into the mechanisms through which obesity may drive cancer pathogenesis.
Materials and Methods
Study population
The study was approved by the Wright State University Institutional Review Board and all methods were performed in accordance with the relevant guidelines and regulations. Whole blood samples were obtained from either the Cooperative Human Tissue Network (CHTN) (15 CRC patients) and Advocate Sherman Hospital (5 CRC patients and 5 lean controls) (Table 1). The informed consent was collected by CHTN and Advocate Sherman Hospital. The DNA methylation data of whole blood samples from obese (n = 10) and lean controls (n = 10) was obtained from a publicly available database (NCBI GEO; accession number GSE85928). RNA-seq datasets for two replicates of HCT116 and DKO cells were obtained from GEO (accession number GSE60106).
DNA extraction, RRBS library preparation and sequencing
Whole genomic DNA was extracted from whole blood using a DNeasy Blood & Tissue Kit (Qiagen, USA) following the manufacturer’s protocol. After checking the quality of the extracted DNA, 500 ng of genomic DNA was digested overnight with Msp1 (New England Biolabs, USA). The sticky ends produced by MspI digestion were filled with CG nucleotides, and 3′A overhangs were added. A DNA library was prepared using NEXTflex Bisulfite-Seq Kit (Bioo Scientific) following a standard procedure. A bisulfite conversion step was performed prior to PCR amplification using the EZ DNA Methylation-Gold kit (Zymo Research Corp.) following the manufacturer’s instructions. All PCR reactions for RRBS were purified using AMPure XP (Beckman Coulter, Brea, USA), and analyzed on a bioanalyzer. Sequencing was performed on the Illumina HiSeq.2500 for a paired-end 2 × 50bp run, with 150 million reads from each direction. Data quality check was done on the Illumina SAV. De-multiplexing was performed with the Illumina Bcl2fastq2 v2.17 program.
Bioinformatics and statistical analysis
The quality of the raw reads was examined with FastQC. The adapter trimming and filtering of the high quality reads was carried out with Cutadapt v1.8.3 and Trim Galore v0.4.0 with the -RRBS option. Quality processed reads were mapped to human genome (hg19) using Bismark assisted by Bowtie2. Before DMC and DMR analyses, methylation calls were filtered by discarding bases with coverage below 5X and bases with more than 99.9th percentile coverage in each sample. CpG sites on sex chromosomes and mitochondrion were excluded from the analyses. Individual DMCs were identified between obesity/CRC and control groups using logistic regression with the R package methylKit. Read coverage was normalized between samples. A minimum of three individuals per group were required for a CpG site to be analyzed. The CpGs with at least 10% methylation difference and a q-value < 0.05 were considered to be differentially methylated. DMRs were determined using the R package eDMR with default parameters. To be considered significant, a DMR needed to contain at least one DMC, three CpG sites, and an absolute mean methylation difference greater than 5%. We annotated the DMRs identified using UCSC Refseq gene models with promoter regions defined as being 2 kb upstream from transcription start site (TSS). CpG islands were defined based on UCSC annotation (http://genome.ucsc.edu/). Functional Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of involved genes were performed using DAVID bioinformatics resources (version 6.8; http://david.abcc.ncifcrf.gov/). The p-value was calculated using the modified Fishers exact test and the GO categories and KEGG pathways were identified as significantly enriched when p value was <0.05. Additional parameters were set to the default values. The Kruskal-Wallis test was used for comparison of DNA methylation levels among all groups, while the Mann-Whitney U test was used for comparison between the groups of subjects. A p value < 0.05 was defined as statistical significance and <0.1 was considered as marginal significance. Raw reads from RNA-seq were trimmed and mapped to human genome (hg19) using Tophat v2.1.1. Gene-level counts were generated using HTSeq v0.6.1 and also validated with Cufflinks v2.2.1. For differential expression analysis, read counts were normalized across libraries using the trimmed mean of M-values (TMM) method implemented in the R package edgeR v3.22.3 and were subsequently transformed to log2-counts per million (log-CPM) and corrected for heteroscedasticity with voom transformation of the R package limma v3.36.2. The log-CPM values of representative genes were visualized as heat map using the heatmap.2 function of the R package gplots v3.0.1 with ‘scale = row’ parameter.
Data Availability
The datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
References
De Pergola, G. & Silvestris, F. Obesity as a major risk factor for cancer. J Obes 2013, 291546 (2013).
Fair, A. M. & Montgomery, K. Energy balance, physical activity, and cancer risk. Methods Mol Biol 472, 57–88 (2009).
Pischon, T., Nothlings, U. & Boeing, H. Obesity and cancer. Proc Nutr Soc 67, 128–145 (2008).
Calle, E. E. & Kaaks, R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer 4, 579–591 (2004).
Dong, Y., et al. Abdominal Obesity and Colorectal Cancer Risk: Systematic Review and Meta-Analysis of Prospective Studies. Biosci Rep (2017).
Ma, Y. et al. Obesity and risk of colorectal cancer: a systematic review of prospective studies. PLoS One 8, e53916 (2013).
Moghaddam, A. A., Woodward, M. & Huxley, R. Obesity and risk of colorectal cancer: a meta-analysis of 31 studies with 70,000 events. Cancer Epidemiol Biomarkers Prev 16, 2533–2547 (2007).
Hidayat, K., Yang, C.M. & Shi, B.M. Body fatness at an early age and risk of colorectal cancer. Int J Cancer (2017).
Ward, H. A. et al. Pre-diagnostic meat and fibre intakes in relation to colorectal cancer survival in the European Prospective Investigation into Cancer and Nutrition. Br J Nutr 116, 316–325 (2016).
Moore, L. D., Le, T. & Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38 (2013).
Holliday, R. & Pugh, J. E. DNA modification mechanisms and gene activity during development. Science 187, 226–232 (1975).
Compere, S. J. & Palmiter, R. D. DNA methylation controls the inducibility of the mouse metallothionein-I gene lymphoid cells. Cell 25, 233–240 (1981).
Baylin, S. B. & Jones, P. A. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 11, 726–734 (2011).
Hellman, A. & Chess, A. Gene body-specific methylation on the active X chromosome. Science 315, 1141–1143 (2007).
Ball, M. P. et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol 27, 361–368 (2009).
Aran, D., Toperoff, G., Rosenberg, M. & Hellman, A. Replication timing-related and gene body-specific methylation of active human genes. Hum Mol Genet 20, 670–680 (2011).
Mohn, F. et al. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 30, 755–766 (2008).
Wong, J. J., Hawkins, N. J. & Ward, R. L. Colorectal cancer: a model for epigenetic tumorigenesis. Gut 56, 140–148 (2007).
Suter, C. M., Martin, D. I. & Ward, R. L. Hypomethylation of L1 retrotransposons in colorectal cancer and adjacent normal tissue. Int J Colorectal Dis 19, 95–101 (2004).
Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 8, 286–298 (2007).
Kuchiba, A. et al. Global methylation levels in peripheral blood leukocyte DNA by LUMA and breast cancer: a case-control study in Japanese women. Br J Cancer 110, 2765–2771 (2014).
Woo, H. D. & Kim, J. Global DNA hypomethylation in peripheral blood leukocytes as a biomarker for cancer risk: a meta-analysis. PLoS One 7, e34615 (2012).
Friso, S. et al. Global DNA hypomethylation in peripheral blood mononuclear cells as a biomarker of cancer risk. Cancer Epidemiol Biomarkers Prev 22, 348–355 (2013).
Barchitta, M., Quattrocchi, A., Maugeri, A., Vinciguerra, M. & Agodi, A. LINE-1 hypomethylation in blood and tissue samples as an epigenetic marker for cancer risk: a systematic review and meta-analysis. PLoS One 9, e109478 (2014).
Mendoza-Perez, J. et al. Genomic DNA Hypomethylation and Risk of Renal Cell Carcinoma: A Case-Control Study. Clin Cancer Res 22, 2074–2082 (2016).
Moore, L. E. et al. Genomic DNA hypomethylation as a biomarker for bladder cancer susceptibility in the Spanish Bladder Cancer Study: a case-control study. Lancet Oncol 9, 359–366 (2008).
Ruike, Y., Imanaka, Y., Sato, F., Shimizu, K. & Tsujimoto, G. Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing. BMC Genomics 11, 137 (2010).
Irizarry, R. A. et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41, 178–186 (2009).
Kanehisa, M., Sato, Y., Furumichi, M., Morishima, K. & Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res 47, D590–D595 (2019).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45, D353–D361 (2017).
Zenonos, K. & Kyprianou, K. RAS signaling pathways, mutations and their role in colorectal cancer. World J Gastrointest Oncol 5, 97–101 (2013).
di Magliano, M. P. & Logsdon, C. D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 144, 1220–1229 (2013).
Bonnans, C., Chou, J. & Werb, Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 15, 786–801 (2014).
Pickup, M. W., Mouw, J. K. & Weaver, V. M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep 15, 1243–1253 (2014).
Lu, P., Weaver, V. M. & Werb, Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196, 395–406 (2012).
Levental, K. R. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906 (2009).
Ng, M. R. & Brugge, J. S. A stiff blow from the stroma: collagen crosslinking drives tumor progression. Cancer Cell 16, 455–457 (2009).
Nakao, A. et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. Embo Journal 16, 5353–5362 (1997).
Katz, M., Amit, I. & Yarden, Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim Biophys Acta 1773, 1161–1176 (2007).
Arvanitis, D. N. & Davy, A. Regulation and misregulation of Eph/ephrin expression. Cell Adhes Migr 6, 131–137 (2012).
Hill, J. O., Wyatt, H. R. & Peters, J. C. Energy balance and obesity. Circulation 126, 126–132 (2012).
Jung, U. J. & Choi, M. S. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci 15, 6184–6223 (2014).
Yang, S. Y., He, X. Y. & Schulz, H. 3-Hydroxyacyl-CoA dehydrogenase and short chain 3-hydroxyacyl-CoA dehydrogenase in human health and disease. FEBS J 272, 4874–4883 (2005).
Rutter, J., Winge, D. R. & Schiffman, J. D. Succinate dehydrogenase - Assembly, regulation and role in human disease. Mitochondrion 10, 393–401 (2010).
Hardwick, J. P., Osei-Hyiaman, D., Wiland, H., Abdelmegeed, M. A. & Song, B. J. PPAR/RXR Regulation of Fatty Acid Metabolism and Fatty Acid omega-Hydroxylase (CYP4) Isozymes: Implications for Prevention of Lipotoxicity in Fatty Liver Disease. PPAR Res 2009, 952734 (2009).
Ameri, K. et al. HIGD1A Regulates Oxygen Consumption, ROS Production, and AMPK Activity during Glucose Deprivation to Modulate Cell Survival and Tumor Growth. Cell Rep (2015).
Zamponi, G. W., Striessnig, J., Koschak, A. & Dolphin, A. C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol Rev 67, 821–870 (2015).
Rhee, I. et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416, 552–556 (2002).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2016. CA Cancer J Clin 66, 7–30 (2016).
SEER Stat Fact Sheets: Colon and Rectum, A.a. (http://seer.cancer.gov/statfacts/html/colorect.html).
Society, A.C. (available at: https://www.cancer.org/cancer/colon-rectal-cancer/detection-diagnosis-staging/detection.html).
Pi-Sunyer, X. The medical risks of obesity. Postgrad Med 121, 21–33 (2009).
Bergstrom, K. S. & Xia, L. Mucin-type O-glycans and their roles in intestinal homeostasis. Glycobiology 23, 1026–1037 (2013).
Bergstrom, K. et al. Defective Intestinal Mucin-Type O-Glycosylation Causes Spontaneous Colitis-Associated Cancer in Mice. Gastroenterology 151, 152–164 e111 (2016).
Kawashima, H. Roles of the gel-forming MUC2 mucin and its O-glycosylation in the protection against colitis and colorectal cancer. Biol Pharm Bull 35, 1637–1641 (2012).
Cornick, S., Tawiah, A. & Chadee, K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers 3, e982426 (2015).
Pascual, J. M. et al. GLUT1 deficiency and other glucose transporter diseases. Eur J Endocrinol 150, 627–633 (2004).
Szablewski, L. Expression of glucose transporters in cancers. Biochim Biophys Acta 1835, 164–169 (2013).
Micucci, C., Orciari, S. & Catalano, A. Hyperglycemia Promotes K-Ras-Induced Lung Tumorigenesis through BASCs Amplification. PLoS One 9 (2014).
Yun, J. et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325, 1555–1559 (2009).
Huang, L. et al. Transcriptional repression of SOCS3 mediated by IL-6/STAT3 signaling via DNMT1 promotes pancreatic cancer growth and metastasis. J Exp Clin Cancer Res 35, 27 (2016).
Zhang, Z. Y. et al. Structural basis for the recognition of Asef by adenomatous polyposis coli. Cell Res 22, 372–386 (2012).
Sieber, O. M., Tomlinson, I. P. & Lamlum, H. The adenomatous polyposis coli (APC) tumour suppressor - genetics, function and disease. Mol Med Today 6, 462–469 (2000).
Massague, J. TGF beta in cancer. Cell 134, 215–230 (2008).
Xu, P. L., Liu, J. M. & Derynck, R. Post-translational regulation of TGF-beta receptor and Smad signaling. Febs Lett 586, 1871–1884 (2012).
Jung, B., Staudacher, J. J. & Beauchamp, D. Transforming Growth Factor beta Superfamily Signaling in Development of Colorectal Cancer. Gastroenterology 152, 36–52 (2017).
Li, L. et al. DNA Methylation in Peripheral Blood: A Potential Biomarker for Cancer Molecular Epidemiology. J Epidemiol 22, 384–394 (2012).
Acknowledgements
This project was supported by Startup funds from Wright State University, the NIH Center of Biomedical Research Excellence on Obesity and Cardiovascular Diseases (P20 GM103527-06), Beginning Grant-in-Aid (11BGIA7710059), and Scientist Development Grant (12SDG12050697) from the American Heart Association.
Author information
Authors and Affiliations
Contributions
H.R., L.D., L.M. and G.M. conceived and designed experiments. H.R. and L.D. performed and analyzed the results. H.R. and L.D. wrote paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dong, L., Ma, L., Ma, G.H. et al. Genome-wide Analysis Reveals DNA Methylation Alterations in Obesity Associated with High Risk of Colorectal Cancer. Sci Rep 9, 5100 (2019). https://doi.org/10.1038/s41598-019-41616-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41616-0
This article is cited by
-
Ketotherapy as an epigenetic modifier in cancer
Reviews in Endocrine and Metabolic Disorders (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
