
Abstract
The cytochrome bo3 quinol oxidase from Vitreoscilla (vbo3) catalyses oxidation of ubiquinol and reduction of O2 to H2O. Data from earlier studies suggested that the free energy released in this reaction is used to pump sodium ions instead of protons across a membrane. Here, we have studied the functional properties of heterologously expressed vbo3 with a variety of methods. (i) Following oxygen consumption with a Clark-type electrode, we did not observe a measurable effect of Na+ on the oxidase activity of purified vbo3 solubilized in detergent or reconstituted in liposomes. (ii) Using fluorescent dyes, we find that vbo3 does not pump Na+ ions, but H+ across the membrane, and that H+-pumping is not influenced by the presence of Na+. (iii) Using an oxygen pulse method, it was found that 2 H+/e− are ejected from proteoliposomes, in agreement with the values found for the H+-pumping bo3 oxidase of Escherichia coli (ecbo3). This coincides with the interpretation that 1 H+/e− is pumped across the membrane and 1 H+/e− is released during quinol oxidation. (iv) When the electron transfer kinetics of vbo3 upon reaction with oxygen were followed in single turnover experiments, a similar sequence of reaction steps was observed as reported for the E. coli enzyme and none of these reactions was notably affected by the presence of Na+. Overall the data show that vbo3 is a proton pumping terminal oxidase, behaving similarly to the Escherichia coli bo3 quinol oxidase.
Similar content being viewed by others

Introduction
Respiring organisms employ the electron transfer chain (ETC) to convert nutrients into the cellular energy currency ATP by a universally conserved mechanism termed oxidative phosphorylation1. The ETC is located in the inner mitochondrial membrane or the plasma membrane of eukaryotes and prokaryotes, respectively. Heme-copper oxidases catalyze the terminal step reducing oxygen to water. This highly exergonic electron transfer is coupled to proton pumping that maintains an electrochemical proton gradient across the membrane utilizing two mechanisms. First, as the site of oxygen reduction is localized in the middle of the membrane, proton and electron uptake from opposite sides of the membrane results in a transmembrane charge separation. Second, many heme-copper oxidases utilize the free energy released during the oxygen reduction to pump protons from the negative (N-) to the positive (P-) side of the membrane, contributing to both the electrical (Δψ) and proton concentration gradient (ΔpH) components2,3. The site where oxygen reduction is catalyzed is highly conserved4. Depending on their electron donor, heme-copper oxidases are divided into either cytochrome c (cyt.c) or quinol oxidases5. The bacterial cytochrome c oxidases typically consist of three core subunits with four redox active sites, termed CuA (sununit II), heme a, CuB, and heme a3 (all in subunit I) that mediate electron transfer during enzymatic turnover. In these oxidases electrons are transferred consecutively from cyt. c via CuA and heme a to the binuclear center (CuB and heme a3), where O2 is bound and reduced to two molecules of water, accompanied by proton uptake and pumping6. In contrast to cytochrome c oxidases, quinol oxidases lack a CuA center and receive the electrons from quinol7. In E. coli, the enzyme consists of four subunits, but all three redox centers, heme b, heme o and CuB, are found in subunit I. The cytochrome bo3 oxidase from E. coli (ecbo3) is thus a quinol dependent oxidase which pumps four protons across the lipid bilayer for each O2 reduced to two H2O8.
These electron and proton-transfer reactions are very fast (ns to ms) and cannot be studied with conventional spectroscopic techniques. Instead, the flow-flash technique has been employed, in which the fully reduced enzyme is inhibited by CO and rapidly mixed with oxygenated buffer. After mixing, the CO ligand is dissociated by means of a short laser flash, allowing the reaction of reduced enzyme with oxygen in a highly synchronized reaction. The electron and proton-transfer reactions are monitored spectrophotometrically at wavelengths specific to the metal cofactors or pH-sensitive dyes, respectively9.
Some organisms utilize a sodium gradient across their membrane to directly or indirectly support ATP synthesis10,11,12,13,14. Na+-translocating F1F0 ATP synthases have been identified in some anaerobic bacteria found in a marine environment (e.g. Ilyobacter tartaricus, Propionigenium modestum, Acetobacter woodii)15,16,17. Additionally, sodium dependent energy-converting enzymes have also been found in bacteria that harbor a H+-translocating F1F0 ATP synthase18. For example, in the Vibrio family, the Na+-translocating NADH-quinone oxidoreductase (Na-NQR) is a primary sodium pump that maintains a sodium-motive force. While the membrane potential can be utilized irrespectively of the translocated ion to synthesize ATP and import nutrients19, Na+/H+ exchangers are thought to control cellular pH and sodium homeostasis and might be able to interconvert ion gradients20,21. In addition to ATP synthases and NQR, complex I of Klebsiella pneumoniae and two heme-copper oxidases have been proposed to transport Na+ ions instead of protons22,23, but see24. One enzyme was purified from Thioalkalivibrio and characterized as a cbb3-type oxidase25,26. The second example is a bo3 quinol oxidase from Vitreoscilla27,28, a gram-negative aerobic bacterium that is known for containing a bacterial hemoglobin29,30. Sequence analysis reveals that the ATP synthase of Vitreoscilla does not use Na+ as its coupling ion (Supplementary Fig. S1) and that the bo3 oxidase shows a high similarity to the H+-pumping enzyme of Escherichia coli31. However, in a series of reports, 22Na-uptake experiments in native Vitreoscilla membranes and with purified enzyme reconstituted into liposomes indicated a Na+-dependent oxidase activity and the ability of the enzyme to pump sodium ions instead of protons32,33. Given the experimental difficulties to differentiate between substrate and pumped protons in single-turnover experiments, an oxidase that pumps sodium ions would represent a unique model enzyme to address important mechanistic questions. Additionally, the high sequence similarity to the enzyme from E. coli would allow to pinpoint the amino-acid residues that are responsible for Na+-pumping. Finally, many pathogenic bacteria rely on the sodium motive force for sodium-dependent drug efflux pumps or motility, making primary sodium pumps attractive drug targets34.
Here, we report on the heterologous expression and purification of the Vitreoscilla bo3 oxidase (vbo3) in E. coli. We studied the purified enzyme using a range of techniques but did not observe any influence of Na+ on the activity of the enzyme nor did we detect any Na+ pumping. Instead, we found that the enzyme pumps protons with a stoichiometry of 1 H+/e− and is functionally indistinguishable from the E. coli counterpart.
Results and Discussion
Sequence analysis
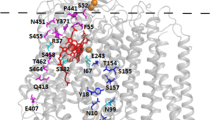
The Vitreoscilla cytochrome cyo operon has been sequenced earlier35, showing that it encodes for subunits I-IV and the protoheme IX farnesyltransferase that is important for heme o biosynthesis36,37. The Vitreoscilla bo oxidase (vbo3) was previously shown to be structurally similar to the E. coli enzyme31. A global sequence alignment of the catalytic subunit I of the two oxidases shows 63% sequence identity and 78% sequence similarity (Supplementary Fig. S2). A homology model of subunit I of the vbo3 oxidase (Fig. 1) was built on the basis of the crystal structure of the ecbo3 (PDB-ID: 1FFT)5 using SwissModel38,39,40. Heme-copper oxidases can be classified into different families2,41,42. Oxidases belonging to the A-type family, including ecbo3, contain two proton translocation pathways (K and D pathway) and have a proton/electron stoichiometry of 2 H+/e−, where 1 H+ is consumed for the chemical reaction and 1 H+ is pumped across the membrane. The K-pathway, which includes residues K362 and Y288 in ecbo3, corresponding to K361 and Y287 in vbo3 (Fig. 1, green sticks), is suggested to deliver 1–2 protons during reduction of the enzyme, while the D-pathway, starting at D135 in ecbo3 (D134 in vbo3) and including E286 in ecbo3 (E285 in vbo3, Fig. 1 dark blue), is used for transfer of the remaining protons during the reductive and oxidative half cycles43. The conserved charged amino acid residues that are involved in proton uptake and/or pumping are listed in Table 1 of the Supplementary Information. All except one key residues identified in subunit I of cytochrome bo3 quinol oxidases are also present in the Vitreoscilla oxidase, the exception being a glutamate (E540) in the E. coli enzyme, which is replaced by an aspartate (D544) in Vitreoscilla5,44,45,46 (Fig. 1, magenta). Residue E540 was suggested to affect overall protein folding in E. coli44. A role in sodium specificity was suggested for the analogue residue D544 in Vitreoscilla47.

Homology model of the Vitreoscilla bo3 oxidase. View from the N-side (left) and side view (right) of the homology model of subunit I from Vitreoscilla bo3 quinol oxidase. The peptide chain is shown as grey cartoon, hemes b and o are depicted as sticks in red, and the copper ion is shown as an orange sphere. Highly conserved key residues in heme-copper oxidases are depicted in stick representation (D-channel residues are dark blue, K-channel residues are green, remaining conserved residues are light blue). D544 of vbo3 oxidase, corresponding to E540 in E. coli, is shown in magenta.
Expression and purification
Starting from genomic DNA, an expression plasmid containing the wild type cyo operon from Vitreoscilla was constructed and the protein was expressed as described in the Material & Methods section. A His-tag at the N-terminus of subunit III allowed purification via Ni-NTA affinity chromatography. SDS PAGE (Supplementary Fig. S3) and mass spectrometry analysis (Table 2 in Supplementary Information) confirmed the presence of all four subunits after affinity purification.
The dithionite reduced minus oxidized spectrum of the heterologously expressed vbo3 was similar to that purified from wild type Vitreoscilla48 and that from the ecbo3 with the typical absorbance peaks at 428 nm, 532 nm and 563 nm43 (Supplementary Fig. S4).
The enzymatic activity of vbo 3 is not affected by Na+ ions
The O2-reduction activity of the purified enzyme was measured using a Clark-type electrode. Electrons were supplied by DTT, using ubiquinone Q1 as an electron mediator (Supplementary Fig. S5). The enzyme turnover in detergent solution was ~250 e−/s, i.e. in the same range as described earlier with the enzyme purified from Vitreoscilla27,28,32,49 and similar to the ecbo3 turnover measured in our lab50.
The O2-reduction activity was inhibited by potassium cyanide (KCN)51,52. The vbo3 enzyme showed maximal activity around pH 8.5 and 50% activity at pH 7 and 9.5 when measured in detergent. No Na+ dependence of the enzymatic activity could be observed at any of the tested pH values (pH 7.5–9.0, the data at pH 8.5 are shown in Fig. 2A). To test whether or not the absence of a membrane could alter the activity, vbo3 was reconstituted into liposomes consisting of either E. coli polar lipids or soybean lecithin. While reconstitution was successful and respiratory control ratios of up to 7 were obtained (Fig. 2B), no influence of 100 mM Na+ (present on both sides of the membrane) on the oxygen-reduction activity was observed (Fig. 2A). In these reconstitution experiments, we used a method established in our lab by mixing liposomes with purified enzyme in the presence of sodium cholate that is subsequently removed by gel filtration (method 1). In these proteoliposomes, about two thirds of the bo3 oxidases are expected to pump outwards with the remaining enzymes pumping in the opposite direction as judged from earlier experiments with the aa3 oxidase53. An alternative method has been described by Verkhovskaya et. al.54 that is supposed to yield a more uniform enzyme orientation, with essentially all enzymes pumping from the inside to the outside (method 2). Using method 2, we aimed to test, if vbo3 pumps Na+ ions across the membrane, following the Na+-concentration in the liposomes with a Na+-specific fluorophore Sodium Green. The dye was entrapped into proteoliposomes containing vbo3 and ecbo3 as well as empty liposomes. However, upon energization of the vbo3 oxidase by DTT/Q1, no signal change was observed. The responsiveness of the dye towards Na+-efflux was confirmed in all three populations by addition of the Na+-specific ionophore monensin in the presence of a Na+-gradient (Fig. 3A).

Oxygen consumption measurements of solubilized and liposome reconstituted vbo3 oxidase. (A) The Na+ dependence of solubilized vbo3 (closed dots) was assessed measuring oxygen consumption using a Clark-type electrode in 50 mM Tris-HCl, pH 8.5, 0.1% DDM and KCl and NaCl concentrations adding up to 300 mM. The reaction was started using DTT and Q1 as electron donor and mediator, respectively. The Na+ dependence of vbo3 reconstituted into liposomes (open dots) was measured in 50 mM Tris-HCl, pH 8.5, 50 mM KCl and NaCl as indicated in the figure. The Na+ concentration was identical inside and outside of the proteoliposomes. (B) The influence of valinomycin and CCCP on vbo3 (open bars) and ecbo3 (closed bars) reconstituted into liposomes was measured in 50 mM HEPES, pH 7.5 and 300 mM KCl. RCRs were calculated as ratio of the activities in the presence and absence of both CCCP and valinomycin (Val/CCCP).

Ion pumping measurements of vbo3 reconstituted into liposomes. (A) Sodium Green, a Na+-sensitive fluorophore and 100 mM NaCl was encapsulated into proteoliposomes containing vbo3 (blue trace) or ecbo3 (orange trace) as well as empty liposomes (grey trace). The proteoliposomes were resuspended in buffer containing 50 mM Hepes, pH 7.5, 100 mM KCl, 1 μM CCCP, 1 mM DTT and quinol dependent oxidase activity was measured by addition of 30 μM Q1. The presence of Na+-gradient was confirmed by addition of 1 μM monensin, a sodium specific ionophore that triggered Na+-efflux. Addition of 100 mM NaCl to the bulk reimports Na+, increasing fluorescence. (B) Inward directed proton pumping was measured in inverted membrane vesicles from Escherichia coli C43 Δcyo cells expressing either vbo3 (blue and green trace) or ecbo3 (orange trance). The vesicles were diluted into buffer containing 50 mM Hepes, pH 7.5, 100 mM KCl (green and orange trace) or 100 mM NaCl (blue trace), 0.2 μM Valinomycin and 2 mM DTT. Proton pumping was followed by ACMA fluorescence and induced by addition of 100 μM Q1. (C) Outwards directed proton pumping was measured with vbo3 proteoliposomes containing 1 mM pyranine in the presence of 100 mM NaCl, 100 mM KCl (blue trace) or in the presence of 200 mM KCl (green trace). Vesicles were diluted into buffer containing 50 mM Hepes, pH 7.5, 100 mM NaCl and 100 mM KCl (blue trace) or 200 mM KCl (green trace), and 2 mM DTT. The reaction was started with addition of 100 μM Q1.
The vbo 3 pumps protons with a 1 H+/e− stoichiometry
The data described above indicate that the vbo3 does not pump Na+ and that the activity of the enzyme is independent of the Na+ concentration. Hence, we investigated proton pumping by vbo3. The approach was applied earlier with Na+-dependent ATP synthases that pump protons in the absence of sodium ions, but switch to sodium pumping if the ion is present at sufficient concentrations55. Proton pumping was investigated by measuring ACMA fluorescence quenching, which reports establishment of a proton gradient (acidic inside). If the enzyme would switch to sodium pumping, proton pumping would be impaired and the fluorescence would not be quenched. When using ACMA to detect proton pumping, the internal volume of the liposome has to become acidified, i.e. the bo3 oxidase has to pump protons inwards, which is opposite to the prevailing orientation during liposome reconstitution (see above). We thus prepared inverted membrane vesicles of E. coli cells lacking chromosomal ecbo3 but expressing vbo3, in which the oxidase is oriented inside-out. As depicted in Fig. 3B, vectorial transport of protons was detected in membranes expressing vbo3 and the ACMA-signal was similar to that obtained with inverted membrane vesicles containing ecbo3. The proton pumping persisted when 100 mM NaCl was added to the outside, indicating that Na+ cannot replace protons as the pumped ion in the vbo3 oxidase. To directly follow in time proton pumping of vbo3, the enzyme was reconstituted into liposomes using method 2 containing the ratiometric pH sensitive fluorophore pyranine. Upon addition of DTT/Q1, proton release from the liposomes was detected as a change in the fluorescence signal (Fig. 3C). Experiments with liposomes containing 100 mM NaCl and liposomes containing the proton pumping ecbo3 oxidase (Supplementary Fig. S6) yielded similar signals, reinforcing the notion that vbo3 is a proton pump. We note that, to our knowledge, the absence of proton pumping in vbo3 has never actually been shown so far. If a preparation of the vbo3 would contain even trace amounts of a proton/sodium exchanger then an analysis of sodium-pumping activity could lead to the conclusion that sodium rather than protons are pumped by the enzyme.
Finally, in order to determine the number of pumped protons per turnover, we assessed proton pumping potentiometrically using a pH electrode as described54,56 (Setup in Fig. 4A). Liposomes containing vbo3 (method 2) or ecbo3 were incubated anaerobically in unbuffered KCl solution at approximately 6.5 to 7.0. The solution contained 3 mM DTT and 100 μM ubiquinone Q1 and residual oxygen was depleted by enzyme turnover. The reaction was started upon addition of a well-defined volume of air-saturated water (~2.5 nmol O2), which leads to proton pumping from the inside to the outside of the liposomes, leading to acidification of the bulk (Fig. 4B, red trace). In addition, upon quinol oxidation, protons are released to the P-side of the membrane, thus also contributing to bulk acidification. Using this method, a ratio of ~2 H+/e− was measured for the enzymes of E. coli and P. denitrificans, corresponding to the release of 1 H+/e− released from quinol oxidation and 1 H+/e− being pumped across the membrane57. Our measurements summarized in Table 1 agree with these findings, showing that both vbo3 and ecbo3 eject around two protons per electron upon enzymatic turnover at pH 6.5 to 7.0. No net proton release to the bulk was observed in the presence of 1 μM CCCP, a membrane proton ionophore that rapidly equilibrates protons from the inside and outside of the liposomes. The very short voltage spike is likely due to an experimental perturbation of the system during injection (Fig. 4B, black trace). Importantly, the presence of 100 mM NaCl did not influence proton pumping and the signals of vbo3 were identical to those obtained with ecbo3 (Table 1).

Determination of H+/e− ratio in ecbo3 and vbo3. (A) Experimental setup of the potentiometric measurements. Shown is a schematic representation of the measurement setup consisting of a pH electrode inserted into a gas-tight measuring chamber. Additions during the experiment are made via a septum using Hamilton syringes. Liposomes containing either reconstituted vbo3 or ecbo3 were incubated in unbuffered 200 mM KCl solution, containing 100 µM Q1 and 2 mM DTT (~pH 6.5–7). The pH of the suspension was followed with a pH electrode connected to a potentiostat. The system was allowed to become fully anaerobic by oxidase turnover as indicated by a stable signal on the potentiostat. An oxygen pulse of 2.5 nmol O2 was applied by adding a defined amount of air-saturated pure water (O2 arrow). The oxygen pulse induced quinol oxidation and proton pumping, leading to an acidification of the bulk that was detected by the pH electrode. The experiment was performed in presence of either 200 mM KCl or 100 mM KCl and 100 mM NaCl (inside and outside of the liposomes). In a control experiment in the presence 1 µM CCCP, no acidification was observed. Each measurement was internally calibrated by the addition of 10 nmol protons from anaerobic 1 mM HCl solution (HCl arrow). (B) Raw traces measured as described in A) using proteoliposomes containing vbo3 in the presence of 200 mM KCl and 100 mM NaCl (red trace) as well as with additional CCCP (black trace).
Single turnover measurements
As mentioned in the introduction section, a motivation to investigate vbo3 was its potential usefulness in single turnover measurements to discriminate substrate from pumped protons. In spite of the negative results described above, we performed a series of single turnover measurements in order to investigate whether the kinetics of internal electron transfer was dependent on the presence of sodium ions. In these experiments, reduced oxidase was incubated with carbon monoxide (CO) and then rapidly mixed with an oxygen-saturated solution. The CO ligand was dissociated synchronously in the entire enzyme population by a laser pulse, which allowed O2 to bind at the catalytic site. Time-resolved spectroscopic measurements revealed discrete electron-transfer steps during reduction of O2 to H2O (see Supplementary Fig. S7 for a scheme of the reaction cycle discussed below). We start with the discussion of the absorbance changes observed at 430 nm, where both hemes contribute to the signal. As seen in Fig. 5A, after the laser flash an unresolved increase in absorbance is observed that corresponds to CO dissociation and oxygen binding to the catalytic site (R→A transition). The signal then rapidly drops with a rate constant (τ) of ~13 µs, reflecting electron transfer (oxidation) from heme b to the catalytic site, corresponding to the A→P transition. The absorbance then slightly increases (τ~50–200 µs, see discussion of the 560 nm data below), reflecting re-reduction of heme b and P→F transition. It is known from measurements with the aa3 oxidases that during P→F transition one proton is taken up to the catalytic site and a second proton is pumped58. Finally, the signal decreases with τ~2 ms, reflecting F→O transition that renders the fully oxidized enzyme. In aa3 oxidases, during this transition one proton is transferred to the catalytic site and one is pumped.

Flow-flash measurements of the reaction of vbo3 with oxygen. (A) Changes of absorbance over time measured at 430 nm upon reaction of reduced vbo3 oxidase with oxygen in the absence of Na+ or in the presence of 100 mM NaCl. The reaction was started by dissociation of CO bound to the catalytic site by a short laser flash. (B) Like A, but absorbance changes at 560 nm were recorded. (C–E) pH dependence of the rates of the different kinetic phases (fit to the traces measured at 430 nm) seen during reaction of vbo3 with O2. Shown are the time constants for the A → P (C), P → F (D) and F → O (E) transitions in presence of 300 mM KCl (blue) or 100 mM NaCl (green).
When the reaction was monitored at 560 nm (Fig. 5B), where essentially only heme b absorbs, the absorbance decreases with τ~20 µs, reflecting the A→P transition. In contrast to the data observed at 430 nm, the re-reduction of heme b during P→F is clearly visible at 560 nm (τ~40 µs), followed in time by the final oxidation in F→O (τ~2 ms). Taken together, the overall oxidative cycle is very similar to that observed in aa3 oxidases and to that observed in the ecbo3 oxidase. Time constants derived from the measurements at different wavelengths are summarized in Table 2. None of these absorbance changes were affected notably by the presence of 100 mM NaCl (Fig. 5A,B).
Finally, a series of flow-flash traces at 430 nm were recorded at different pH values in the presence and absence of 100 mM NaCl (Fig. 5C–E). As observed with other H+-dependent and H+-pumping oxidases4,59, the A→P transition was not affected by the pH, while pH dependencies were observed for the proton pumping steps P→F and F→O (Fig. 5D,E). Again, the presence of NaCl did not majorly influence the kinetics of any of these reactions. Taken together, all single turnover measurements further support the data from multiple turnover measurements that vbo3 is a true proton pumping quinol oxidase.
In contrast to cytochrome c oxidases that harbor four redox-active sites, quinol oxidases without a bound quinol can only store three electrons, which is not sufficient for a complete enzyme turnover that requires 4 electrons. In contrast, if quinol is present, the reduced enzyme harbors five electrons, which is sufficient to completely reduce oxygen to water leaving one electron in the oxidase. With our preparation, we found that addition of external quinone Q1 was not necessary to observe a complete oxidative cycle (Supplementary Fig. S8). However, when the intrinsically bound quinone was replaced by added inhibitor HQNO, the reaction was impaired after delivery of the third electron (P→F transition). From these data, we conclude that our preparation of vbo3 was co-purified with a bound ubiquinone5,60.
Concluding Remarks
We describe the expression and functional characterization of a quinol bo3 oxidase from Vitreoscilla. In contrast to earlier findings, we did not detect any Na+-dependent oxidase activity nor did we observe any sodium pumping. Instead, we show that vbo3 is a proton pumping oxidase that is functionally very similar to the closely related enzyme of E. coli. The rate of quinol-driven oxygen reduction of vbo3 is in the same range as the rate determined for ecbo3. Quantitative proton pumping experiments revealed that the enzyme pumps 1 H+/e−, similar to quinol oxidase from E. coli and e.g. the mitochondrial cytochrome c oxidase. In addition, internal electron-transfer reactions displayed similar time constants to those observed earlier for ecbo3 and these time constants were insensitive to addition of Na+. The existence of a Na+-pumping quinol oxidase would have been an exciting addition to the already impressive collection of heme copper oxidases, but such an enzyme seems to require more substantial sequence differences than found between the bo3 oxidases of E. coli and Vitreoscilla.
Materials and Methods
If not stated otherwise, chemicals were purchased from Sigma-Aldrich.
Protein expression and purification
The plasmid pET-17b harbouring the Vitreoscilla cyoABCDE operon, cloned from Vitreoscilla genomic DNA (kind gift from Prof. Benjamin Stark, Illinois Institute of Technology) using the following primers: fw: 5′-ATAGATATACATATGAAGCAGATGATTCAGGTCTTATCTTTTATCACG-3′, Re: 5′-ATAGAGAGCAAGCTTTAATCAAAAATAAATATGCGGCAACAAATGTTTCAC-3′, and containing a N-terminal His9-tag to cyoC and an E. coli optimized RBS, was transformed into E. coli C43 ΔcyoABDCE cells. Cells were grown aerobically in LB containing 100 μM CuSO4 and 1 mM MgSO4, pH 7.5 at 37 °C at 220 rpm. Protein expression was induced at OD600nm = 0.5 by adding 2 mM IPTG. The temperature was lowered to 30 °C and the cells were kept overnight at 220 rpm. Cells were harvested and disrupted passing them three times through a Maximator HPL6 (Maximator AG) at 30–35kPsi. Cell debris was collected by centrifugation (20’000 × g, 20 min, 4 °C) and membranes were collected by ultracentrifugation (180’000 × g, 90 min, 4 °C). The membrane fraction (10 mg/ml total protein) was solubilized in 50 mM HEPES (Santa Cruz Biotechnology), pH 8.5, 20 mM imidazole, 2% (w/v) DDM (Glycon Biochemicals) for 2 h at 4 °C. vbo3 was purified by Ni-NTA-affinity chromatography using a 1 ml HisTrap FF column (GE Healthcare Life Sciences). After loading, the column was washed with 20 column volumes 50 mM HEPES, pH 8.5, 20 mM imidazole, 0.1% DDM, followed by 5 column volumes 50 mM HEPES, pH 8.5, 60 mM imidazole, 0.1% DDM. The protein was eluted with 50 mM HEPES, pH 8.5, 500 mM imidazole, 0.1% DDM, until the eluate was colorless. Imidazole concentration was reduced by repeated washing steps using a concentrator tube (Amicon, 100 kDa). The concentrated vbo3 (~25 uM) was flash-frozen with LN2 and kept at −80 °C.
Activity measurements
Steady state measurements of oxygen reduction activity of vbo3 was determined using a Clark-type oxygraph (Hansatech). The reaction was started by addition of 1 mM DTT to a solution containing 50 mM HEPES, pH 7.5, 0.1% DDM, 100 µM Q1. Different NaCl or KCl concentration were present as described in the text.
Membrane protein reconstitution into liposomes
Method 1: Reconstitution using cholate and P10 desalting column as described53. Briefly, the desired amount of E. coli polar lipids (Avanti Polar Lipids) dissolved in chloroform (20 mg/ml) was dried under a stream of argon gas and kept under vacuum in a desiccator overnight. The next morning, the lipid film was suspended in buffer (50 mM HEPES, pH 8.5, 300 mM KCl) to a concentration of 5 mg/ml. The suspension was sonicated on ice (2 min and 30 pulse in total, 30 seconds pulse, 30 seconds break, 40% amplitude) to form unilamellar liposomes. To this suspension, 0.6% sodium cholate (Panreac AppliChem) was added followed by addition of ~0.5 μM vbo3 or ecbo3. The sample was incubated at 25 °C for 20 minutes while being inverted from time to time. Excess cholate was removed by means of a Centripure P10 column (emp biotech).
Method 2: Reconstitution using the Bio-beads protocol after Verkovskaya et. al.54. Soybean lecithin, 90% (Alfa Aesar) or E. coli polar lipid extract was resuspended in 200 mM HEPES, pH 7.5, 1.6% n-octyl-β-D-glucopyranosideside (Anatrace) to 8 mg/ml. After sonication (2 min and 30 pulse in total, 30 seconds pulse, 30 seconds break, 40% amplitude), 0.75 to 1 μM vbo3 or ecbo3 was added and incubated at 25 °C on a slowly turning wheel for 15 minutes was followed by addition 80 mg/ml (wet weight) Bio-Beads (Biorad). After 30 minutes, another portion of 80 mg/ml Bio-Beads was added and the sample was incubated for another hour.Subsequently, 160 mg/ml Bio-Beads were added and incubated for 2 more hours, followed by another 160 mg/ml and 2 more hours incubation. The beads were allowed to settle and the supernatant was transferred to an ultracentrifuge tube and diluted 1:10 with 200 mM KCl solution. Proteoliposomes were collected by ultracentrifugation (180’000 × g, 90 min, 4 °C) and the pellet was carefully rinsed with 200 mM KCl before resuspension with in the starting volume with 200 mM KCl. Liposomes containing sodium were rinsed and resuspended with solution containing 100 mM NaCl and 100 mM KCl instead and allowed to equilibrate overnight. All measurements were performed within 24 hours of preparation without a notable loss of activity.
Proteoliposome Na+- and H+-pumping assays
For Na+-pumping assays, vbo3 was reconstituted into E. coli polar extract (Avanti Polar Lipids) using method 2. The sodium specific dye Sodium Green TMA salt (Thermo Fisher) was encapsulated inside the liposomes during reconstitution at 10 μM. Non-encapsulated dye was removed during the last ultracentrifugation step followed by a CentriPure MINI Spin Column Desalt Z-50. Sodium pumping was measured following the fluorescence using the Cary Eclipse fluorescence spectrophotometer using 507 nm and 532 nm as excitation and emission wavelength respectively. The reaction was started by adding 2 mM DTT to a stirred suspension containing vbo3, ecbo3 or empty proteoliposomes, 100 μM Q1, 1 μM CCCP in 50 mM Hepes, pH 7.5, followed by additions of 1 μM monensin and the indicated amount of NaCl or KCl.
Proton pumping was measured using pyranine as pH sensitive dye essentially as described before61. For this, vbo3 was reconstituted into asolectin liposomes (method 2), and 1 mM pyranine (Thermo Fisher Scientific) was encapsulated inside the proteoliposomes during reconstitution in the presence or absence of 100 mM NaCl. The ratio of the fluorescent signals at 406/510 and 460/510 decreases upon addition of Q1, indicating an increase in pH inside the liposomes Proton pumping was also assessed potentiometrically using a micro pH electrode VWR. Liposomes (method 2)were prepared and diluted in either 200 mM KCl, pH 7.4, or 100 mM KCl, 100 mM NaCl, pH 7.4, to compare the pumping in presence and absence of sodium. After liposome addition, 0.2 μM Valinomycin and 2 mM DTT were added. The reaction chamber was sealed to eliminate gas-exchange with the atmosphere. The measurement was then performed as described in54. Briefly, after sealing the chamber the initial pH was adjusted to 6.5–7.0 by adding 10 mM HCl or 10 mM NaOH. Enzyme turnover was started by adding 100 μM Q1 and the system was allowed to become anaerobic (seen as stabilization of the pH and the voltage read by the potentiostat). The pH was noted again and an oxygen pulse corresponding to 2.5 nmol O2 was supplied by adding 10 μl of air saturated pure water. After the baseline was reached again, the system was internally calibrated by addition of 10 μl 1 mM anaerobic HCl.
Inverted Membrane Vesicle H+-pumping assay
Inward directed proton pumping was assessed in inverted membrane vesicles from E. coli C43 Δcyo cells expressing either ecbo3 or vbo3. Membranes were prepared as described under protein expression and purification using a Maximator HPL6 (Maximator AG) followed by ultracentrifugation steps. The membranes were resuspended in 50 mM HEPES, pH 7.5 and either 100 mM KCl or 100 mM NaCl. The inverted membrane vesicles were equilibrated for equal proton and sodium concentrations inside and outside overnight. Proton pumping was followed measuring 9-amino-6-chloro-2-methoxyacridine (ACMA) fluorescence at 418 nm and 483 nm as excitation and emission wavelength, respectively. Membranes were diluted in 50 mM HEPES, pH 7.5 and either 100 mM KCl or 100 mM NaCl, 2 μM ACMA, 0.2 μM Valinomycin, and 2 mM DTT were supplied. After recording the baseline, turnover was started by adding 100 μM Q1. The pH gradient was dissipated by addition of 30 mM NH4Cl from a 1 M stock solution.
Single turnover measurements
Flow- flash measurements for single turnover were performed as described earlier62,63. Briefly, purified samples were diluted to 10 μM (in presence and absence of 100 mM NaCl) and transferred to a Thunberg cuvette and the atmosphere was exchanged for N2 on a vacuum line. The sample was fully reduced by addition of 10 μM hexamine ruthenium and 2 mM sodium ascorbate from the sidearm of the cuvette. Reduction state of the enzyme was followed spectrophotometrically. Complete reduction occurred after about 8 hours of incubation. After that, the atmosphere was exchanged for CO on a vacuum line. Using a locally modified stopped-flow apparatus (Applied Photophysics), the reduced and CO-blocked protein sample was mixed 1:5 with oxygen-saturated buffer (~1.2 mM O2). After a delay of 200 ms, CO was dissociated from the catalytic site by a short laser pulse (~10 ns laser flash (λ = 532 nm, Nd YAG-laser, Quantel) to allow oxygen binding. Changes in absorbance were recorded over time at the indicated wavelengths. For the measurements with N-oxo-2-heptyl-4-Hydroxyquinoline (HQNO), 25 μM HQNO was added to the sample and incubated for 15–30 minutes at room temperature. The data were fitted to a kinetic model using the ProK software from Applied Photophysics, UK.
References
Marreiros, B. C. et al. Exploring membrane respiratory chains. Biochim. Biophys. Acta - Bioenerg. 1857, 1039–1067 (2016).
Lee, H. J., Reimann, J., Huang, Y. & Ädelroth, P. Functional proton transfer pathways in the heme-copper oxidase superfamily. Biochim. Biophys. Acta - Bioenerg. 1817, 537–544 (2012).
Brzezinski, P. & Ädelroth, P. Design principles of proton-pumping haem-copper oxidases. Curr. Opin. Struct. Biol. 16, 465–472 (2006).
Yoshikawa, S. & Shimada, A. Reaction mechanism of cytochrome c oxidase. Chem. Rev. 115, 1936–89 (2015).
Abramson, J. et al. The structure of the ubiquinol oxidase from Escherichia coli and its ubiquinone binding site. Nat. Struct. Mol. Biol. 7, 910–7 (2000).
Ferguson-Miller, S. & Babcock, G. T. Heme/Copper Terminal Oxidases. Chem. Rev. 96, 2889–2908 (1996).
Puustinen, A., Finel, M., Haltia, T., Gennis, R. B. & Wikstrom, M. Properties of the two terminal oxidases of Escherichia coli. Biochemistry 30, 3936–3942 (1991).
Choi, S. K. et al. Location of the Substrate Binding Site of the Cytochrome bo 3 Ubiquinol Oxidase from Escherichia coli. J. Am. Chem. Soc. 139, 8346–8354 (2017).
Einarsdóttir, Ó., Funatogawa, C., Soulimane, T. & Szundi, I. Kinetic studies of the reactions of O2 and NO with reduced Thermus thermophilus ba 3 and aa 3 using photolabile carriers. Biochim. Biophys. Acta - Bioenerg. 1817, 672–679 (2012).
Mulkidjanian, A. Y., Galperin, M. Y., Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Evolutionary primacy of sodium bioenergetics. Biol. Direct 3, 13 (2008).
Skulachev, V. P. Membrane-linked energy transductions. Bioenergetic functions of sodium: H+ is not unique as a coupling ion. Eur. J. Biochem. 151, 199–208 (1985).
Steuber, J. et al. Central role of the Na+-translocating NADH:quinone oxidoreductase (Na+-NQR) in sodium bioenergetics of Vibrio cholerae. Biol. Chem. 395, 1389–99 (2014).
Dimroth, P. & von Ballmoos, C. ATP synthesis by decarboxylation phosphorylation. Results Probl. Cell Differ. 45, 153–84 (2008).
Dimroth, P. & Cook, G. M. Bacterial Na+ - or H+ -coupled ATP synthases operating at low electrochemical potential. Adv. Microb. Physiol. 49, 175–218 (2004).
Reidlinger, J. & Müller, V. Purification of ATP synthase from Acetobacterium woodii and identification as a Na+-translocating F1F0-type enzyme. Eur. J. Biochem. 223, 275–83 (1994).
Neumann, S., Matthey, U., Kaim, G. & Dimroth, P. Purification and properties of the F1F0 ATPase of Ilyobacter tartaricus, a sodium ion pump. J. Bacteriol. 180, 3312–6 (1998).
Laubinger, W. & Dimroth, P. Characterization of the Na+-stimulated ATPase of Propionigenium modestum as an enzyme of the F1F0 type. Eur. J. Biochem. 168, 475–80 (1987).
Barquera, B. The sodium pumping NADH:quinone oxidoreductase (Na+-NQR), a unique redox-driven ion pump. J. Bioenerg. Biomembr. 46, 289–98 (2014).
Tokuda, H. & Unemoto, T. Characterization of the respiration-dependent Na+ pump in the marine bacterium Vibrio alginolyticus. J. Biol. Chem. 257, 10007–14 (1982).
Dibrov, P. A., Lazarova, R. L. & Skulachev, V. P. The sodium cycle. II. Na+-coupled oxidative phosphorylation in Vibrio alginolyticus cells. 850, 458–465 (1986).
Dibrov, P. A., Lazarova, R. L., Skulachev, V. P. & Verkhovskaya, M. L. A study on Na+ -coupled oxidative phosphorylation: ATP formation supported by artificially imposed ΔpNa and ΔpK in Vibrio alginolyticus cells. J. Bioenerg. Biomembr. 21, 347–57 (1989).
Gemperli, A. C., Dimroth, P. & Steuber, J. The respiratory complex I (NDH I) from Klebsiella pneumoniae, a sodium pump. J. Biol. Chem. 277, 33811–7 (2002).
Gemperli, A. C., Dimroth, P. & Steuber, J. Sodium ion cycling mediates energy coupling between complex I and ATP synthase. Proc. Natl. Acad. Sci. USA 100, 839–44 (2003).
Bertsova, Y. V. & Bogachev, A. V. The origin of the sodium-dependent NADH oxidation by the respiratory chain of Klebsiella pneumoniae. FEBS Lett. 563, 207–12 (2004).
Muntyan, M. S. et al. Cytochrome cbb 3 of Thioalkalivibrio is a Na+-pumping cytochrome oxidase. Proc. Natl. Acad. Sci. USA 112, 7695–700 (2015).
Murali, R., Yildiz, G. G., Daldal, F. & Gennis, R. B. Stoichiometry of proton pumping by the cbb 3-type oxygen reductase in whole cells of Rhodobacter capsulatus at pH 7 is about 0.5 H+ per electron. Proc. Natl. Acad. Sci. USA 109, E2144; author reply E2145 (2012).
Efiok, B. J. S. & Webster, D. A. Respiratory-driven sodium electrical potential in the bacterium, Vitreoscilla. Biochemistry 29, 4734–4739 (1990).
Efiok, B. J. S., Webster, D. A. & Sodium-coupled, A. T. P. synthesis in the bacterium. Vitreoscilla. Arch. Biochem. Biophys. 292, 102–106 (1992).
Wakabayashi, S., Matsubara, H. & Webster, D. A. Primary sequence of a dimeric bacterial haemoglobin from Vitreoscilla. Nature 322, 481–3.
Stark, B. C., Dikshit, K. L. & Pagilla, K. R. The Biochemistry of Vitreoscilla hemoglobin. Comput. Struct. Biotechnol. J. 3, e201210002 (2012).
Georgiou, C. D., Cokic, P., Carter, K., Webster, D. A. & Gennis, R. B. Relationship between membrane-bound cytochrome o frome Vitreoscilla and that of Escherichia coli. 933, 179–183 (1988).
Efiok, B. J. S. & Webster, D. A. A cytochrome that can pump sodium ion. 173, 370–375 (1990).
Park, C., Moon, J. Y., Cokic, P. & Webster, D. A. Na+-translocating cytochrome bo terminal oxidase from Vitreoscilla: Some parameters of its Na+ pumping and orientation in synthetic vesicles. Biochemistry 35, 11895–11900 (1996).
Hase, C. C., Fedorova, N. D., Galperin, M. Y. & Dibrov, P. A. Sodium Ion Cycle in Bacterial Pathogens: Evidence from Cross-Genome Comparisons. Microbiol. Mol. Biol. Rev. 65, 353–370 (2001).
Hwang, K. W. et al. Isolation, sequencing, and characterization of the cytochrome bo operon from Vitreoscilla. DNA Seq. 14, 53–59 (2003).
Kim, S. K., Stark, B. C. & Webster, D. A. Evidence that Na+-pumping occurs through the D-channel in Vitreoscilla cytochrome bo. Biochem. Biophys. Res. Commun. 332, 332–8 (2005).
Saiki, K., Mogi, T. & Anraku, Y. Heme o biosynthesis in Escherichia coli: the cyoE gene in the cytochrome bo operon encodes a protoheme IX farnesyltransferase. Biochem. Biophys. Res. Commun. 189, 1491–7 (1992).
Biasini, M. et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, 252–258 (2014).
Arnold, K., Bordoli, L., Kopp, J. & Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 (2006).
Guex, N., Peitsch, M. C. & Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 30, 162–173 (2009).
Pereira, M. M., Santana, M. & Teixeira, M. A novel scenario for the evolution of haem-copper oxygen reductases. Biochim. Biophys. Acta 1505, 185–208 (2001).
Hemp, J. & Gennis, R. B. In Bioenergetics: Energy Conservation and Conversion (eds Schäfer, G. & Penefsky, H. S.) 1–31, https://doi.org/10.1007/400_2007_046 (Springer Berlin Heidelberg, 2008).
Kobayashi, K., Tagawa, S. & Mogi, T. Electron Transfer Processes in Subunit I Mutants of Cytochrome bo Quinol Oxidase in Escherichia coli. Biosci. Biotechnol. Biochem. 73, 1599–1603 (2009).
Kawasaki, M., Mogi, T. & Anraku, Y. Substitutions of charged amino acid residues conserved in subunit I perturb the redox metal centers of the Escherichia coli bo-type ubiquinol oxidase. J. Biochem. 122, 422–9 (1997).
Minagawa, J., Mogi, T., Gennis, R. B. & Anraku, Y. Identification of heme and copper ligands in subunit I of the cytochrome bo complex in Escherichia coli. J. Biol. Chem. 267, 2096–104 (1992).
Hellwig, P., Yano, T., Ohnishi, T. & Gennis, R. B. Identification of the Residues Involved in Stabilization of the Semiquinone Radical in the High-Affinity Ubiquinone Binding Site in Cytochrome bo 3 from Escherichia coli by Site-Directed Mutagenesis and EPR Spectroscopy †. Biochemistry 41, 10675–10679 (2002).
Chung, Y. T., Stark, B. C. & Webster, D. A. Role of Asp544 in subunit I for Na+ pumping by Vitreoscilla cytochrome bo. Biochem. Biophys. Res. Commun. 348, 1209–1214 (2006).
Georgiou, C. D. & Webster, D. A. Purification and partial characterization of the membrane-bound cytochrome o(561,564) from Vitreoscilla. Biochemistry 26, 6521–6 (1987).
Georgiou, C. D., Cokic, P., Carter, K., Webster, D. A. & Gennis, R. B. Relationships between membrane-bound cytochrome o from Vitreoscilla and that of Escherichia coli. Biochim. Biophys. Acta 933, 179–83 (1988).
Berg, J., Block, S., Höök, F. & Brzezinski, P. Single Proteoliposomes with E. coli Quinol Oxidase: Proton Pumping without Transmembrane Leaks. Isr. J. Chem. 1–10, https://doi.org/10.1002/ijch.201600138 (2017).
Parashar, A., Venkatachalam, A., Gideon, D. A. & Manoj, K. M. Cyanide does more to inhibit heme enzymes, than merely serving as an active-site ligand. Biochem. Biophys. Res. Commun. 455, 190–193 (2014).
Van Heyningen, W. E. The inhibition of respiration by cyanide. Biochem. J. 29, 2036–9 (1935).
von Ballmoos, C., Biner, O., Nilsson, T. & Brzezinski, P. Mimicking respiratory phosphorylation using purified enzymes. Biochim. Biophys. Acta 1857, 321–31 (2016).
Verkhovskaya, M. L. et al. Glutamic acid 286 in subunit I of cytochrome bo 3 is involved in proton translocation. Proc. Natl. Acad. Sci. USA 94, 10128–31 (1997).
Laubinger, W. & Dimroth, P. The sodium ion translocating adenosinetriphosphatase of Propionigenium modestum pumps protons at low sodium ion concentrations. Biochemistry 28, 7194–8 (1989).
Basilio, D. & Accardi, A. A Proteoliposome-Based Efflux Assay to Determine Single-molecule Properties of Cl− Channels and Transporters. J. Vis. Exp. 1–9, https://doi.org/10.3791/52369 (2015).
Verkhovskaya, M., Verkhovsky, M. & Wikström, M. pH dependence of proton translocation by Escherichia coli. J. Biol. Chem. 267, 14559–62 (1992).
Brzezinski, P. Redox-driven membrane-bound proton pumps. Trends Biochem. Sci. 29, 380–387 (2004).
Johansson, A.-L. et al. Proton-transport mechanisms in cytochrome c oxidase revealed by studies of kinetic isotope effects. Biochim. Biophys. Acta 1807, 1083–94 (2011).
Puustinen, A., Verkhovsky, M. I., Morgan, J. E., Belevich, N. P. & Wikstrom, M. Reaction of the Escherichia coli quinol oxidase cytochrome bo 3 with dioxygen: the role of a bound ubiquinone molecule. Proc. Natl. Acad. Sci. USA 93, 1545–1548 (1996).
Verkhovskaya, M., Knuuti, J. & Wikström, M. Role of Ca2+ in structure and function of Complex I from Escherichia coli. Biochim. Biophys. Acta - Bioenerg. 1807, 36–41 (2011).
Ädelroth, P., Ek, M. S., Mitchell, D. M., Gennis, R. B. & Brzezinski, P. Glutamate 286 in cytochrome aa 3 from Rhodobacter sphaeroides is involved in proton uptake during the reaction of the fully-reduced enzyme with dioxygen. Biochemistry 36, 13824–9 (1997).
Brändén, M. et al. On the role of the K-proton transfer pathway in cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 98, 5013–8 (2001).
Acknowledgements
We thank Prof. Pia Ädelroth (Stockholm University) for helpful discussions. The work was supported by grants from the Swiss National Science Foundation (No. 153351) and the Knut and Alice Wallenberg Foundation (KAW, Sweden).
Author information
Authors and Affiliations
Contributions
C.v.B., P.B. and S.G. designed functional experiments. S.G. performed all experiments, and together with C.v.B. and P.B., analyzed the data. C.v.B., S.G. and P.B. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Graf, S., Brzezinski, P. & von Ballmoos, C. The proton pumping bo oxidase from Vitreoscilla. Sci Rep 9, 4766 (2019). https://doi.org/10.1038/s41598-019-40723-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40723-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
