
Abstract
Stylosanthes guianensis (Aubl.) Sw. is an economically important pasture and forage legume in tropical regions of the world. Genetic improvement of the crop can be enhanced through marker-assisted breeding. However, neither single nucleotide polymorphism (SNP) markers nor SNP-based genetic linkage map has been previously reported. In this study, a high-quality genetic linkage map of 2572 SNP markers for S. guianensis is generated using amplified-fragment single nucleotide polymorphism and methylation (AFSM) approach. The genetic map has 10 linkage groups (LGs), which spanned 2226.6 cM, with an average genetic distance of 0.87 cM between adjacent markers. Genetic mapping of quantitative trait loci (QTLs) for important agronomic traits such as yield-related and nutritional or quality-related traits was performed using F2 progeny of a cross between a male-sterile female parent TPRC1979 and male parent TPRCR273 with contrasting phenotypes for morphological and physiological traits. A total of 30 QTLs for 8 yield-related traits and 18 QTLs for 4 nutritional or quality-related traits are mapped on the linkage map. Both the high-quality genetic linkage map and the QTL mapping for important agronomic traits described here will provide valuable genetic resources for marker-assisted selection for S. guianensis.
Similar content being viewed by others
Introduction
Important agronomic traits are generally inherited in a quantitative way, and marker-assisted selection (MAS) is very useful for tracing elite alleles that control these important, complex traits thus accelerating the breeding process1. However, the construction of a high-density genetic linkage map is the prerequisite for genetic dissection of a target trait through quantitative trait locus (QTL) analysis. Moreover, high-resolution genetic linkage map is an essential and powerful tool for positional cloning of genes, scaffold sequence anchoring and genome assembly, and comparative genomic analysis2.
Although many types of DNA markers have been developed since 1980s, the application of random amplification of polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP) and sequence-related amplified polymorphism (SRAP) has been limited due to their dominant inheritance, low abundance and low transferability3. Simple sequence repeat (SSR) markers are also unqualified for the construction of high-density genetic linkage map because of their limited number and coverage and lacking sequence information, despite having advantages over aforementioned marker types such as co-dominant inheritance, locus specificity and reproducibility3. Single nucleotide polymorphisms (SNPs) are by far the most abundant and stable mutations identified between related DNA molecules, and uniformly distributed in the genomes4. The traditional tools for development of SNP markers are generally expensive and labor-intensive. The advances in next-generation sequencing (NGS) promoted the development of SNP markers and facilitated the high throughput SNP genotyping at low cost5,6,7,8. Several NGS-based approaches have been developed to identify SNPs, such as restriction site associated DNA sequencing (RAD-seq)9, genotyping-by-sequencing (GBS)10, 2b-RAD11, specific length amplified fragment sequencing (SLAF-seq)12, double digest RAD (ddRAD)13 and amplified-fragment single nucleotide polymorphism and methylation (AFSM)14. These technologies have been successfully applied to construct high-resolution genetic linkage maps for many important crops15,16,17,18,19.
The genus Stylosanthes, belonging to the tribe Aeschynomeneae, subtribe Stylosanthinae and the family Fabaceae, comprises 48 species native to tropical, subtropical and temperate regions of America, Africa and Southeast Asia, mainly South America20,21. Stylosanthes (stylo) is currently used as cut-and-carry feed for livestock, wasteland reclamation, fallow crops and hay22,23. Several species of stylo such as S. guianensis, S. hamata, S. scabra, S. fruticosa, S. seabrana, are considered as economically important tropical pasture legumes due to their high productivity, wide adaptability, potential in improving soil fertility (through nitrogen fixation, reclaiming degraded wastelands, and water and soil conservation) and extensive use in a range of agricultural systems22. Of these, S. guianensis is the most widespread perennial herbaceous Stylosanthes species in Australia, China, India and many other tropical countries of Africa, Southeast Asia and South America24,25. S. guianensis, a diploid species (2n = 20) with great morphological diversity26, has a mixed mating system with predominance of autogamy, and the outcrossing rate is estimated as 26%27.
Although genetic linkage maps of an interspecific cross between S. hamata and S. scabra28, and of S. scabra29 had been reported with RAPD markers of low polymorphism, to date, high-density genetic linkage map for S. guianensis is not available. Even the literature on the development and application of molecular markers for S. guianensis is still relatively scarce. Microsatellites or simple sequence repeats (SSRs) have been developed to study the genetic diversity and population structure of S. guianensis30,31,32, and to determine the mating system of S. guianensis. Sequence-related amplified polymorphism (SRAP) has been used to select the true hybrids of S. guianensis33. The generation of a high-density genetic linkage map is critical step toward providing the framework for stylo genetic improvement through MAS. However, due to the non-availability of co-dominant markers of high level polymorphism, high-density linkage map for S. guianensis has not been generated successfully.
AFSM represents a quick and simple SNP discovery based on the use of two restriction enzyme pairs (EcoRI-MspI and EcoRI-HpaII) and next-generation sequencing. It has been successfully applied to construct high-density linkage map for cassava (Manihot esculanta Crantz), proving that it is an efficient and cost-effective for high-throughput strategy for de novo SNP genotyping of large genomes14. Here, we describe the development of the first high-density linkage map using AFSM approach and QTL mapping for important agronomic traits in S. guianensis using the genome-wide composite interval mapping (GCIM). These resources will help to promote genetic dissection of important agronomic traits and subsequent MAS in accelerating genetic improvement for S. guianensis.
Materials and Methods
Plant materials and DNA extraction
A F2 population of 202 individuals was produced by self-pollinating a F1 hybrid derived from the cross between S. guianensis var. TPRC1979 (female) and var. TPRCR273 (male) with contrasting phenotypes for morphological and physiological traits34. Blocks for F2 population, F1 and their parents were established in a randomized complete block design with plant and row spacing of 2 m at the base of teaching and research (Danzhou Campus), Tropical Agriculture and Forestry Institute, Hainan University in 2015. Total genomic DNA was extracted from the young fully-expanded leaves of the parents, F1 and F2 progenies with the DNeasy 96 Plant Kit (Qiagen), and the two technical replicates were pooled for each individual. DNA was quantified using a NanoDrop 2000C spectrophotometer (ND2000; Thermo Fisher Scientific, USA), and DNA concentrations were normalized to 100 ng/μL. DNA quality and integrity were measured by electrophoresis on 1.0% agarose gels.
AFSM library construction, sequencing and genotyping
AFSM library construction, sequencing and genotyping followed the previously established protocol14. The genomic DNA of each sample was digested with two restriction enzyme pairs, EcoRI-MspI and EcoRI-HpaII (New England BioLabs Inc., Ipswich, MA) and ligated to barcode adapters and methylation adapters. Restriction fragments from each library were then amplified, and the resulting PCR products ranging from 250 bp to 500 bp in size were isolated to construct EcoRI-MspI library and EcoRI-HpaII library. Then paired-end sequencing was conducted on an Illumina HiSeq2500 system (Illumina Inc., San Diego, CA). Sequenced reads were processed using custom Perl scripts (http://afsmseq.sourceforge.net/), and the filtered reads were used for SNP identification using the TASSEL-GBS pipeline35 and VCFtools_v0.1.9 (http://vcftools.sourceforge.net/)36.
Genetic linkage map construction
Bi-allelic SNPs identification and genetic linkage map construction with JoinMap 4.137 were performed using procedures described by Xia et al.14. After the evaluation of all pairs of tags for the presence of at least five reads, bi-allelic SNPs (indels and methylation polymorphism sites) were identified by querying the filtered tags for pairs of sequences with the following requirements: identical in at least five reads; present in >50% of the individuals; passed a Fisher’s exact test for independence; fit to the expected Mendelian segregation ratio as calculated with the chisquare test (χ2) for P < 0.01; possessed a recombination frequency of <0.4; had AFSM markers specific to the female or male parent that fit to a 1:1 segregation ratio in addition to shared AFSM markers that fit to a 3:1 segregation ratio in the F1 population. The parent-specific AFSM markers, which segregated at a 1:1 ratio in the population, were set to the lm × ll (marker in the female parent) and nn × np (marker in the male parent) configuration, respectively. The AFSM markers that were present in both parents and segregated at a 3:1 ratio in the population were labeled as hk × hk (marker present in both parents) configuration. The Kosambi mapping function was used for map distance estimation, and the “Ripple” function was employed to confirm the orders of markers within each linkage group.
Phenotypic assessment of morphological and physiological indexes
Agronomic traits of F2 population were determined with three replicates for each item, including 10 yield-related indexes such as fresh weight (FW), dry weight (DW), the fresh weight: dry weight ratio (FW/DW), plant height (PH), primary branch number (PBN), plant width (PW), the maximum length of branches (LB), leaf length (LL), leaf width (LW) and the leaf length: leaf width ratio (LL/LW), and 8 nutritional or quality-related indexes such as the contents of crude protein, crude fiber, crude fat, crude ash, calcium, phosphate, potassium and magnesium. Plants propagated from cuttings of F1 and F2 individuals were used to repeat the experiments. Phenotypic evaluation and data analysis were carried out with the methods described by Ding et al.34.
QTL analysis and mapping
The GCIM38, QTL.gCIMapping 3.1, was applied to map QTLs. Permutation tests were executed to determine the statistical significance of LOD threshold for each trait with the number of permutations set at 10,000. logarithm of the odds (LOD) peaks exceeding LOD threshold (>2.5) were identified as QTL, and based on the location of its LOD peak and the surrounding region, the position of each QTL was determined.
Results
Genetic linkage mapping
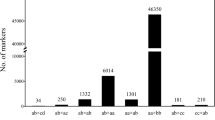
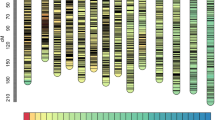
A total of 223,011 SNPs were identified with AFSM approach. After filtering, the low-quality SNPs with coverage less than one third of mapping population and the SNPs showing a statistically significant deviation from Mendelian segregation were removed, and a final set of 13,661 high-quality SNP markers were evaluated for subsequent genetic linkage mapping. Excluding the unlinked and co-segregated SNP markers, we constructed the linkage map with 2572 markers distributed across 10 linkage groups (LGs). The resulting map had a total length of 2226.6 cM and an average interlocus distance of 0.87 cM (Fig. 1, Table 1, Table S1). The longest LG (LG9) contained 249 markers spanning 317.3 cM and the shortest LG (LG2) contained 273 markers spanning 121 cM, with an average genetic distance between successive markers being 1.27 cM and 0.44 cM, respectively. The highest-density LG (LG1) consisted of 600 markers spanning 162.6 cM, with an average marker interval being 0.41 cM.

Genetic linkage map constructed for Stylosanthes guianensis using SNP markers. LG: linkage group. Scale on the left indicated map distance in centimorgan. Black horizontal lines indicate marker positions on the map.
Evaluation of the phenotypic data
On the basis of the phenotypic data34, it was revealed that two parents were clearly different in terms of the agronomic traits, and the phenotypic values for each trait of segregation population exhibited wide range, with the coefficient of variation (CV) ranging from 8.31% (LL) to 46.19% (FW) for the yield-related traits and from 10.32% (crude ash content) to 34.01% (crude fat content) for nutritional or quality-related traits (Tables S2, S3 and S4). Transgressive segregation in both directions was observed for all traits. All traits exhibited normal distributions based on the testing for normality with Shapiro-Wilk test, suggesting they are quantitative traits (Tables S5 and S6).
Correlation analysis showed that highly significant positive correlation existed between the FW and other yield-related traits (Table S7). Crude protein content had highly significant positive correlation with P content, but had highly significant negative correlation with crude fiber content and not any obvious correlation with other nutritional traits (Table S8). Crude fiber content was significantly positively correlated with crude fat content but significantly negatively correlated with crude ash content and P content. Ca content had significant positive correlations with P content and Mg content but significant negative correlation with K content.
QTL mapping analysis of agronomic traits
A total of 30 QTLs, significant at a LOD score of >2.5, were identified for 8 yield-related traits including PW, LB, PH, LL, LW, PBN, FW/DW and LL/LW (Table 2). Eighteen QTLs controlling four nutritional or quality-related traits (crude protein content, crude ash content, calcium content, and potassium content) were mapped on the linkage map (Table 3).
Five QTLs for PW were identified on four LGs, namely, 2 on LG5, 1 on LG6, 1on LG7 and 1 on LG9 respectively, and explained 1.9–9.5% of the total phenotypic variance at a LOD score of 3.04–9.62. Twelve QTLs for LB were mapped to three LGs, namely, 6 QTLs on LG4, 3 QTLs on LG5 and 3 QTLs on LG9, and the phenotypic variances explained by these QTLs varied from 1.1% to 4.3% at the LOD scores of 5.64–65.82. Seven QTLs for PH were detected on five LGs, namely, 1 on LG2, 2 on LG3, 1 on LG4, 1 on LG5, 1 on LG7 and 1 on LG8, explaining 1.8–6.6% of the phenotypic variance at a LOD score of 2.52–4.65. Only one QTL related to LL were detected on LG8, accounting for 1.3% of the phenotypic variance at a LOD score of 2.51. Only one QTL for LW were identified on LG8, accounting for 4.9% of the phenotypic variance at a LOD score of 4.65. Only one QTL for PBN were identified on LG4, accounting for 56.2% of the phenotypic variance at a LOD score of 5.37, and had the largest contribution to the trait’s variance. Two QTLs for FW/DW were distributed across two LGs, namely, 1 on LG8 and 1 on LG10, explaining 4.2–5.0% of the phenotypic variance at a LOD score of ranging from 5.28 to 6.39. Only one QTL for LL/LW identified on LG9, explained 1.8% of the total phenotypic variance at a LOD score of 2.57.
Four QTLs for calcium content were distributed on LG2 (1), LG5 (1), LG6 (1), and LG9 (1), explaining 1.4–5.7% of the phenotypic variation with LOD scores ranging from 2.51 to 4.80. Ten QTLs for crude protein content were distributed across 6 LGs, namely, LG3 (2), LG4 (2), LG5 (2), LG7 (1), LG8 (1) and LG9 (2). The phenotypic variance explained by these QTLs ranged from 1.5% to 8.7%, with LOD scores of 2.58–8.18. Three QTLs associated with potassium content were mapped on LG2 and LG6, accounting for 2.7–4.3% of the phenotypic variation at the LOD score of 4.74–5.14. For crude ash content, only one QTL was detected on LG4, with phenotypic variation explained of 6.9% at the LOD score of 2.50.
Discussion
Construction of a high-density genetic map can provide a valuable resource for understanding the basis of important complex agronomic traits in S. guianensis. For S. guianensis, the major hurdles in constructing useful genetic map included the lack of the highly polymorphic, co-dominant molecular markers, and the difficulty in artificial emasculation and hybridization. Taking the advantages of the AFSM technology, a cost-effective and improved high-throughput SNP discovery approach which is better suited for non-model species14, and with a mapping population created with a male-sterile female parent (S. guianensis var. TPRC1979) newly found in China, we successfully developed the first high-density genetic linkage map for S. guianensis with 2572 polymorphic SNP markers, which spanned 2226.6 cM with an average distance of 0.87 cM between the flanking markers. With the extensive construction of genetic linkage maps, many excellent software programs have been developed such as FsLinkageMap by Tong et al.39 for constructing high-density genetic linkage maps in some outcrossing plant species, especially in forest trees. S. guianensis is a perennial herbaceous species with predominance of autogamy, and the parents, S. guianensis var. TPRC1979 (female) and var. TPRCR273 (male), could be regarded as inbred lines, thus the linkage map construction in this study was performed with the JoinMap 4.137. S. guianensis has 10 chromosomes in its haploid genome and current genetic map developed in this study is composed of 10 LGs, suggesting that the number of LGs is consistent with haploid chromosome number, and provides adequate genome coverage.
Kazan et al.28 developed a linkage map containing a total of 44 RAPD loci based on an interspecific cross between Stylosanthes hamata and S. scabra. Thumma et al.29 reported that a genetic linkage map of S. scabra consisting of 120 RAPD markers had been constructed, with the total genome length of 1406 cM and an average interval of 12 cM. However, these maps with average marker spacing above 5 cM could not be considered as the high-density maps. In addition, RAPD markers are not only low polymorphic and anonymous (their nucleotide sequences are not known) but also difficult to be transferred to new populations due to their dominant nature.
A high-density genetic map is principally applicable for fine-mapping of QTLs for important agronomic traits. Despite the importance of the agronomic traits such as yield-related and nutritional or quality-related traits, few QTL studies for these traits have been reported due to the lack of high-density genetic maps. Here, for the first time, we mapped the QTLs for the important agronomic traits on the basis of the high-density genetic map we obtained. In total, 30 QTLs for 8 yield-related traits (PW, LB, PH, LL, LW, PBN, FW/DW and LL/LW) and 18 QTLs controlling 4 quality-related traits (crude protein content, crude ash content, calcium content, and potassium content) were detected on the linkage map. Preliminary mapping QTLs with the MapQTL 5.040 using both interval mapping (IM) and multiple QTL model (MQM) mapping generated many problematic results such as two QTLs in the same marker interval, the total of phenotypic variation explained by those QTLs more than 100%, and too many major effective QTLs detected. These issues were addressed very well by the GCIM developed by Wen et al.38, which is a more powerful method for the detection of closely linked and small-effect QTLs. Next step is to validate the QTL mapping in different environments and/or genetic backgrounds and in relatively large mapping populations.
In summary, we have constructed the first high-density genetic map for S. guianensis, which provides a platform for mapping QTLs and cloning genes and comparative genomic study. Once confirmed, the localization of these major QTLs on the linkage map based on next generation sequencing methods will be valuable resources for QTL cloning and genetic improvement by pyramiding favorable alleles into the improved cultivars.
References
Collard, B. C. Y., Jahufer, M. Z. Z., Brouwer, J. B. & Pang, E. C. K. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 142, 169–196, https://doi.org/10.1007/s10681-005-1681-5 (2005).
Jones, N., Ougham, H., Thomas, H. & Pasakinskiene, I. Markers and mapping revisited: finding your gene. New Phytol. 183, 935–66, https://doi.org/10.1111/j.1469-8137.2009.02933.x (2009).
Wang, L. et al. Construction of a high-density linkage map and fine mapping of QTL for growth in Asian seabass. Sci Rep. 5, 16358, https://doi.org/10.1038/srep16358 (2015).
Qiu, G. F. et al. A second generation SNP and SSR integrated linkage map and QTL mapping for the Chinese mitten crab Eriocheir sinensis. Sci Rep. 7, 39826, https://doi.org/10.1038/srep39826 (2017).
Davey, J. W. et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 12, 499–510, https://doi.org/10.1038/nrg3012 (2011).
Nielsen, R., Paul, J. S., Albrechtsen, A. & Song, Y. S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 12, 443–451, https://doi.org/10.1038/nrg2986 (2011).
Kumar, S., Banks, T. W. & Cloutier, S. SNP discovery through next generation sequencing and its applications. Int. J. Plant Genomics 2012, 831460, https://doi.org/10.1155/2012/831460 (2012).
Poland, J. A. & Rife, T. W. Genotyping-by-sequencing for plant breeding and genetics. Plant Genome 5, 92–102, https://doi.org/10.3835/plantgenome2012.05.0005 (2012).
Baird, N. A. et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. Plos One 3, e3376, https://doi.org/10.1371/journal.pone.0003376 (2008).
Elshire, R. J. et al. A robust, simple Genotyping-by-Sequencing (GBS) approach for high diversity species. Plos One 6, e19379, https://doi.org/10.1371/journal.pone.0019379 (2011).
Wang, S., Meyer, E., Mckay, J. K. & Matz, M. V. 2b-RAD: a simple and flexible method for genome-wide genotyping. Nat Methods 9, 808–810, https://doi.org/10.1038/nmeth.2023 (2012).
Sun, X. et al. SLAF-seq: an efficient method of large-scale De novo SNP discovery and genotyping using high-throughput sequencing. Plos One 8, e58700, https://doi.org/10.1371/journal.pone.0058700 (2013).
Dacosta, J. M. & Sorenson, M. D. Amplification biases and consistent recovery of loci in a double-digest RAD-seq protocol. Plos One 9, e106713, https://doi.org/10.1371/journal.pone.0106713 (2014).
Xia, Z., Zou, M., Zhang, S., Feng, B. & Wang, W. AFSM sequencing approach: a simple and rapid method for genome-wide SNP and methylation site discovery and genetic mapping. Sci. Rep. 4, 7300, https://doi.org/10.1038/srep07300 (2014).
Hussain, W. et al. Genotyping-by-sequencing derived high-density linkage map and its application to QTL mapping of flag leaf traits in bread wheat. Sci Rep. 7, 16394, https://doi.org/10.1038/s41598-017-16006-z (2017).
Hu, X. H. et al. High-density genetic Map Construction and identification of QTLs controlling oleic and linoleic acid in peanut using SLAF-seq and SSRs. Sci Rep. 8, 5479, https://doi.org/10.1038/s41598-018-23873-7 (2018).
Huang, H. et al. High-density genetic map construction and comparative genome analysis in asparagus bean. Sci Rep. 8, 4836, https://doi.org/10.1038/s41598-018-23173-0 (2018).
Bai, B. et al. Developing genome-wide SNPs and constructing an ultrahigh-density linkage map in oil palm. Sci Rep. 8, 691, https://doi.org/10.1038/s41598-017-18613-2 (2018).
Liu, T. et al. Construction of the first high-density genetic linkage map of Salvia miltiorrhiza using specific length amplified fragment (SLAF) sequencing. Sci Rep. 6, 24070, https://doi.org/10.1038/srep24070 (2016).
Costa, N. M. S. & Ferreira, M. B. Some Brazilian species of Stylosanthes. In: Stace, H. M. & Edye L. A. (eds) The biology and agronomy of Stylosanthes, (Sydney: Academic Press), 53–101 (1984).
Costa, N. Revisão do género Stylosanthes. PhD Thesis, Universidade Te´cnica de Lisboa, Portugal (2006).
Chandra, A., Pathak, P. S. & Bhatt, R. K. Stylosanthes research in India: Prospects and challenges ahead. Cur. Sci. 90, 915–921 (2006).
Chandra, A. Biotechnology of Stylosanthes. In: Jain, S. M. & Gupta, S. D. (eds) Biotechnology of Neglected and Underutilized Crops. Media Dordrecht: Springer, 217–242, https://doi.org/10.1007/978-94-007-5500-0_10 (2013).
Williams, R. J., Reid, R., Schultze-Kraft, R., Costa, N. M. & Thomas, B. D. Natural distribution of Stylosanthes. In: Stace, H. & Edye, L. (eds). The biology and agronomy of Stylosanthes. Sydney: Academic Press, 73–101 (1984).
Vieira, M. L. C., Aguiar-Perecin, M. L. R. & Martins, P. S. A cytotaxonomic study in twelve Brazilian taxa of Stylosanthes Sw., Leguminosae. Cytologia 58, 305–311, https://doi.org/10.1508/cytologia.58.305 (1993).
Stace, H. & Cameron, D. Cytogenetics and the evolution of Stylosanthes. In: Stace, H. & Edye, L. (eds). The biology and agronomy of Stylosanthes. Sydney: Academic Press, 49–72 (1984).
Santos-Garcia, M. O., Resende, R. M. S., Chiari, L., Zucchi, M. I. & de Souza, A. P. Mating systems in tropical forages: Stylosanthes capitata Vog. and Stylosanthes guianensis (Aubl.) Sw. Euphytica 178, 185–119, https://doi.org/10.1007/s10681-010-0293-x (2011).
Kazan, K., Manners, J. M. & Cameron, D. F. Inheritance of random amplified polymorphic DNA markers in an interspecific cross in the genus Stylosanthes. Genome 36, 50–56 (1993).
Thumma, B. R. et al. Identification of causal relationships among traits related to drought resistance in Stylosanthes scabra using QTL analysis. J. Exp. Bot. 355, 203–214 (2001).
Vander Stappen, J., Weltjens, I. & Volckaert, G. Microsatellite markers in Stylosanthes guianensis. Mol. Ecol. 8, 514–517 (1999).
Santos, M. O. et al. Isolation and characterization of microsatellite loci in the tropical forage legume Stylosanthes guianensis (Aubl.) Sw. Conserv. Genet. Resour. 1, 43–46, https://doi.org/10.1007/s12686-009-9010-2 (2009).
Santos-Garcia, M. O. et al. Identification of Stylosanthes guianensis varieties using molecular genetic analysis. AoB Plants 2012, pls001, https://doi.org/10.1093/aobpla/pls001 (2012).
Huang, C. Q., Liu, G. D., Bai, C. J., Wang, W. Q. & Tang, J. Application of SRAP markers in the identification of Stylosanthes guianensis hybrids. Mol. Bio. Rep. 41, 5923–5929, https://doi.org/10.1007/s11033-014-3467-0 (2014).
Ding, Y.-C., Tang, Y.-Q., Ding, Z.-T., Liu, F. & Yao, J. Establishment of Stylosanthes genetic family and analysis of its genetic variability. Mole. Plant Breed. 14, 997–1002, https://doi.org/10.13271/j.mpb.014.000997 (2016).
Glaubitz, J. C. et al. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. Plos One 9, e90346, https://doi.org/10.1371/journal.pone.0090346 (2014).
Danecek, P. et al. The variant call format and VCFtools. Bioinform. 27, 2156–2158, https://doi.org/10.1093/bioinformatics/btr330 (2011).
Van Ooijen, J. W. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genetics Res. 93, 343–349, https://doi.org/10.1017/S0016672311000279 (2011).
Wen, Y. J., et al An efficient multi-locus mixed model framework for the detection of small and linked QTLs in F2. Brief Bioinform, https://doi.org/10.1093/bib/bby058 (2018)
Tong, C., Zhang, B. & Shi, J. A hidden Markov model approach to multilocus linkage analysis in a full-sib family. Tree Genet Genomes 6, 651–662, https://doi.org/10.1007/s11295-010-0281-2 (2010).
Van Ooijen, J. W. JoinMap @ 4, Software for the calculation of genetic linkage maps in experimental populations, Kyazma B.V.,Wageningen, Netherlands (2006).
Acknowledgements
This work was supported by grants from Hainan Natural Science Foundation 317015 (to Y.Q.T.). This work was also partially supported by National Natural Science Foundation of China Nos. 31360261 and 31860676 (to Y.Q.T.), 31560021 and 31772887 (to Z.L.), and Science and Technology Program of Hainan Province ZDYF2017020 (to Z.L.).
Author information
Authors and Affiliations
Contributions
Y.-Q.T. was responsible for designing this study and drafting the manuscript; Z.-Q.X. and Z.-T.D. carried out the map construction and QTL mapping; Y.-C.D. performed the phenotyping of the mapping populations and data analysis; Z.L. and X.M. participated in data analysis; J.-P.L. co-conceptualized the project and contributed to drafting the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tang, YQ., Xia, ZQ., Ding, ZT. et al. Construction of a high-density linkage map and QTL mapping for important agronomic traits in Stylosanthes guianensis (Aubl.) Sw.. Sci Rep 9, 3834 (2019). https://doi.org/10.1038/s41598-019-40489-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40489-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
