
Abstract
Serine proteinase inhibitors (serpins), typically fold to a metastable native state and undergo a major conformational change in order to inhibit target proteases. However, conformational lability of the native serpin fold renders them susceptible to misfolding and aggregation, and underlies misfolding diseases such as α1-antitrypsin deficiency. Serpin specificity towards its protease target is dictated by its flexible and solvent exposed reactive centre loop (RCL), which forms the initial interaction with the target protease during inhibition. Previous studies have attempted to alter the specificity by mutating the RCL to that of a target serpin, but the rules governing specificity are not understood well enough yet to enable specificity to be engineered at will. In this paper, we use conserpin, a synthetic, thermostable serpin, as a model protein with which to investigate the determinants of serpin specificity by engineering its RCL. Replacing the RCL sequence with that from α1-antitrypsin fails to restore specificity against trypsin or human neutrophil elastase. Structural determination of the RCL-engineered conserpin and molecular dynamics simulations indicate that, although the RCL sequence may partially dictate specificity, local electrostatics and RCL dynamics may dictate the rate of insertion during protease inhibition, and thus whether it behaves as an inhibitor or a substrate. Engineering serpin specificity is therefore substantially more complex than solely manipulating the RCL sequence, and will require a more thorough understanding of how conformational dynamics achieves the delicate balance between stability, folding and function required by the exquisite serpin mechanism of action.
Similar content being viewed by others
Introduction
Over 1,500 serpins have been identified to date. Inhibitory family members typically fold to a metastable native state that undergoes a major conformational change (termed the stressed [S] to relaxed [R] transition) central for the protease inhibitory mechanism1,2. The S to R transition is accompanied by a major increase in stability. The archetypal serpin fold is exemplified by α1-antitrypsin (α1-AT), a single domain protein consisting of 394 residues, which folds into 3 β-sheets (A → C) and 9 α-helices (A → I) that surround the central β-sheet scaffold3. The reactive center loop (RCL) protrudes from the main body of the molecule and contains the scissile bond (P1 and P1’ residues), which mediates α1-AT’s inhibitory specificity against the target protease, neutrophil elastase (HNE).
The inhibitory mechanism of serpins is structurally well understood1. Briefly, a target protease initially interacts with and cleaves the RCL of the serpin. However, following RCL cleavage, but prior to the final hydrolysis of the acyl enzyme intermediate, the RCL inserts into the middle of the serpin’s β-sheet A to form an extra strand1,4. The opening of β-sheet A is controlled by the shutter and the breach regions5. Since the protease is still covalently linked to the P1 residue, the process of RCL insertion results in the translocation of the protease to the opposite end of the molecule. In the final complex, the protease active site is distorted and trapped as the acyl enzyme intermediate1,6.
In certain circumstances the serpin RCL can spontaneously insert, either partially (delta conformation), or fully (latent conformation) into the body of the serpin molecule without being cleaved7. Both latent and delta conformations are considerably more thermodynamically stable than the active, native state although they are inactive as protease inhibitors. Folding to the latent conformation is thought to occur via a late, irreversible folding step that is accessible from the native or a highly native-like state8,9. As such, transition to the latent state can be triggered by perturbations to the native state via small changes in solution conditions such as temperature or pH6,10,11, or by spontaneous formation over long time scales12,13.
Human α1-AT is an extremely potent inhibitor of its target protease HNE, with a rate of association (kass) 6 × 107 M−1 s−1, forming a serpin−protease complex that is stable for several days14,15. The metastable nature of α1-AT is required to facilitate the large conformational change required for its inhibitory function, and the rate of RCL insertion into β-sheet A is the main determinant of whether the acyl linkage between serpin and protease is maintained or disrupted. If RCL insertion is rapid, the inhibitory pathway proceeds. If the RCL insertion is too slow, the serpin becomes a substrate; the de-acylation step of the protease’s catalytic mechanism is complete and cleavage of the P1−P1’ bond occurs without protease inhibition. The cleaved, de-acylated RCL still inserts into the body of the serpin, resulting in an inactive inhibitor16.
Two regions of the RCL appear to govern inhibitory function and specificity. The first, a highly-conserved hinge region (resides P15−P9) consisting of short chain amino acids, facilitates RCL insertion into the A β-sheet. Mutations in the hinge region result in the serpin becoming a substrate rather than an inhibitor17. The second region is the P1 residue, thought to determine specificity towards a protease. Serpins with a P1 arginine (e.g. antithrombin III) are known to target proteases of the coagulation cascade, including thrombin and Factor Xa18,19. In α1-AT, mutation of P1 methionine to arginine (the Pittsburgh mutation), changes the specificity from HNE to thrombin, resulting in a bleeding disorder20.
Given the importance of the RCL, it has been the focus of previous attempts aimed at altering serpin specificity, via mutation of RCL residues or swapping RCL sequences between serpins. Chimeric serpins have been made between plasminogen activator inhibitor-1 (PAI-1) and antithrombin-III (ATIII)21,22, α1-AT and antithrombin-III23,24, α1-AT and ovalbumin25, and alpha1-antichymotrypsin (ACT) and α1-AT15,26,27. In all cases, specificity could only be transferred partially, as each chimera has a reduced second-order rate constant and a higher SI to a target protease in comparison to the original serpin. The most effective chimera produced, without a cofactor, was ACT with P3−P3’ of α1-AT. This chimera achieved a stoichiometry of inhibition (the number of moles of serpin required to inhibit one mole of protease (SI)) of 1.4 and a second-order rate constant (k’/[I]) of 1.1 x 105 M−1 s−1, two orders of magnitude slower than that of α1-AT15. Therefore, it is highly likely that the determinants of specificity are more complex than the RCL region alone, and other regions may play a role, for example exosite interactions in the serpin−protease complex22,28,29,30.
In previous work, we designed and characterized conserpin, a synthetic serpin that folds reversibly, is functional, thermostable and resistant to polymerization31. Conserpin was designed using consensus engineering, using a sequence alignment of 212 serpin sequences and determining the most frequently occurring amino acid residue at each position. Since it is thermostable and easier to produce in recombinant form, it is ideally suited as a model in protein engineering studies. Conserpin shares 59% sequence identity to α1-AT, with 154 residue differences scattered throughout the structure. Its RCL sequence is sufficiently different from all other serpins such that it no longer resembles an RCL of any serpin with a known target protease. A recent study that investigated the folding pathway of conserpin engineered the P7-P2’ sequence of α1-AT into its RCL32. The resulting conserpin/α1-AT chimera inhibits chymotrypsin with an SI of 1.46, however, no SI was calculated against HNE. The chimera forms a weak complex with HNE that is detectable using SDS-PAGE, however, the majority of the serpin molecules are cleaved without complex formation.
In this study, we have exploited the unique folding characteristics of conserpin and employ it as a model serpin with which to investigate the determinants of specificity. We investigated the effect of replacing the RCL of conserpin with the corresponding sequence from α1-AT on inhibitory specificity towards HNE. Here, the chimera molecule, called conserpin-AATRCL, remains thermostable, yet despite possessing the RCL sequence of α1-AT, specificity against HNE was not restored to the extent of α1-AT. Structural analysis and molecular dynamics simulations indicate that specificity is also governed by other, complex factors involving RCL dynamics, and surface electrostatics of regions external to the RCL.
Results
Biophysical and functional characterisation of a conserpin/α1-AT chimera
With the aim of changing the specificity of conserpin to that of α1-AT, a conserpin/α1-AT chimera was previously produced32, where 9 residues within the RCL (P7-P2’) were swapped with the corresponding residues from α1-AT (Fig. 1A). The resulting chimera, conserpin-AATRCL (379 aa) has a 61% sequence identity with α1-AT (148 residue differences). Conserpin-AATRCL was expressed in E. coli and purified from the soluble fraction by affinity and size exclusion chromatography as described previously31.

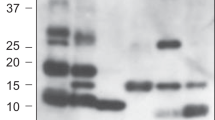
Stability and inhibitory activity of conserpin-AATRCL. (A) RCL sequence alignment indicating which residues of conserpin were replaced with the corresponding residues in α1-AT; (B) Variable temperature thermal melt of conserpin-AATRCL, heating to 95 °C (black line) and cooling to 35 °C (red line), measured by CD at 222 nm; (C) Spectral scan before (black line) and after (red line) variable temperature thermal melt; (D) Variable temperature thermal melt in the presence in 2 M GdnHCl (heating to 95 °C; black line, cooling: red line); (E) Inhibitory activity assay and (F) SI against trypsin (n = 3); (G) A cropped SDS-PAGE showing a serpin:protease complex formed between HNE and AAT, but less complex formed between HNE and conserpin-AATRCL. From left to right: 1. Molecular weight markers (kDa); 2. α1-AT alone; 3. 1:1 ratio of α1-AT: HNE; 4. 2:1 ratio of α1-AT:HNE; 5. HNE alone; 6. conserpin-AATRCL alone; 7. 1:1 ratio of conserpin-AATRCL:HNE; 8. 2:1 ratio of conserpin-AATRCL:HNE. The full length SDS-PAGE gel is presented in Fig. S1.
We first investigated the biophysical properties of conserpin-AATRCL to ensure that swapping the RCL did not alter them. The majority of serpins irreversibly unfold upon heating with a midpoint temperature transition (Tm) of ~55–65 °C33,34,35. Using variable temperature far-UV circular dichroism (CD) to measure the thermostability, conserpin-AATRCL was heated from 35 to 95 °C at a rate of 1 °C/min, and upon reaching 95 °C, minute changes in signal were observed. Following a subsequent 1 °C/min decrease in temperature from 95 to 35 °C, minute changes in signal was observed (Fig. 1B). In addition, far-UV spectral scans before and after thermal unfolding showed minute differences in the signals, suggesting the absence of a large heat-induced conformational change (Fig. 1C). Complete unfolding was only achieved in the presence in 2 M guanidine hydrochloride (GdnHCl) with a Tm of 72.2 ± 0.1 °C. Upon cooling from 95 to 35 °C, no precipitation was observed (Fig. 1D). Thus, high thermostability is consistent with the parent conserpin molecule31 and indicates that incorporation of the α1-AT RCL does not reduce the thermostability of the conserpin scaffold.
We have previously shown conserpin to be a poor inhibitor of trypsin in comparison to α1-AT (SI = 1.8 vs 1.0 respectively)31. Engineering the RCL sequence of α1-AT into conserpin improves the SI against trypsin from 1.8 to 1.64 (conserpin-AATRCL SI = 1.64 ± 0.2 n = 3; Fig. 1E,F). Conserpin-AATRCL, like conserpin, after denaturation and refolding was active against trypsin (SI = 2.0). Importantly, conserpin-AATRCL does not inhibit HNE, the protease target of α1-AT. An SI could not be calculated, as there was residual HNE activity after 30-minute incubation, even with at a 2:1 serpin:protease molar ratio.
If the inhibitory pathway of serpin proceeds faster than the substrate pathway, then the SI will be close to 1. If, however, the inhibitory mechanism is too slow and the substrate pathway occurs, the SI is greater than 136. SDS-PAGE using 1:1 and 2:1 serpin: protease molar ratios reveals a faint complex between conserpin-AATRCL and HNE, but also showed a large amount of cleaved species compared to the complex formation between α1-AT and HNE (Fig. 1G, Fig. S1). Since we observe that conserpin-AATRCL is able to inhibit trypsin, and is still able to transition to the latent state upon heating, we hypothesized that the RCL mutations do not prevent its insertion into β-sheet A. We therefore sought to investigate the structure and dynamics of conserpin-AATRCL in order to identify other factors contributing to its inability to inhibit HNE.
The role of electrostatics in the formation of a serpin:protease complex
To understand if there are any structural changes caused by modifying the RCL, we determined the x-ray crystal structure of conserpin-AATRCL in the native state (Table S1). The overall structure of conserpin-AATRCL is identical to that of conserpin—a structural alignment reveals a root mean square deviation (RMSD) of 0.2 Å across all Cα atoms. Like conserpin and indeed many other serpins, the RCL of conserpin-AATRCL is too flexible to be modelled into the electron density. Therefore, all further analyses were performed with the RCL modelled using the structure of wildtype α1-AT (PDB ID: 3NE437).
Effective serpin inhibition of a protease must involve association to form an encounter complex followed by formation of a stereospecific, high-affinity complex that positions the RCL of the serpin to engage with the protease active site. Given the failure to engineer the RCL for α1-AT specificity and inhibition, we reasoned that surface electrostatics may contribute to the formation and stability of a serpin:protease complex and thus protease inhibition. The electrostatic potential surfaces of conserpin, conserpin-AATRCL and α1-AT differ in several regions. Both conserpin-AATRCL and α1-AT feature a large electropositive surface centred around the loop connecting strands 2 and 3 of β-sheet B (s2B and s3B) (Fig. 2B,C). In conserpin-AATRCL, this patch extends to encompass the D-helix, P9−P1 of the RCL, and strand 2 of β-sheet C (s2C)−helix H (Fig. 2E). The corresponding region on α1-AT is much smaller, covering a region under the RCL, some residues of s1B and its connecting loop to helix G, s4B and s5B (Fig. 2F).

Structure and electrostatics of conserpin-AATRCL. (A,D) X-ray crystal structure of native state conserpin-AATRCL represented as a cartoon. The breach and shutter regions are marked with black broken circles. (B–F) A comparison of electrostatic potential surfaces (blue = +ve, red = −ve) of (B,E) conserpin-AATRCL and (C,F) α1-AT. Both conserpin-AATRCL and α1-AT feature a large electropositive surface centred around the loop connecting strands 2 and 3 of β-sheet B (s2B and s3B) (B,C). A large surface patch between helix D and the RCL, highlighted with yellow broken circles, has a generally positive potential in conserpin-AATRCL (E), and negative potential in α1-AT (F).
A second difference is seen on the top surface of the serpins, directly beneath the RCL. Differences between α1-AT and conserpin-AATRCL—particularly in s2C, s3C, and the loop between s3A and s3C—lead to a large difference in charge on the surface beneath P9−P1 (Fig. 3A,B). In conserpin-AATRCL (and conserpin), this region has a large electropositive potential, while the corresponding region in α1-AT is more neutral in charge (Fig. 3A,B).

Electrostatic potential surfaces of the RCL differs between conserpin-AATRCL and α1-AT. While we have grafted the α1-AT RCL (cartoon) from P7−P2’ onto conserpin (surface), the electrostatic surface potential between conserpin-AATRCL and α1-AT differs beneath the RCL. (A) In conserpin-AATRCL, the region below the RCL contains a large electropositive potential, while in α1-AT (B), the corresponding region is more neutral in charge. (C) ConSURF conservation scores for the serpin superfamily, mapped onto the surface of α1-AT as colours from forest green (highly conserved) to brick red (highly variable). This depicts poor conservation (red) of residues 201−202 and 223−225 of α1-AT, suggesting that these residues may be responsible for contributing to protease specificity within the serpin family.
Functional requirements of an inhibitory serpin’s RCL provide selective pressures on its sequence. In inhibitory serpins, the sequence of the RCL must correspond to the specificity of its target proteases7, maintain a linear, mobile structure in the stressed/native state, and still remain capable of insertion into highly conserved regions in β-sheet A post-cleavage (an example is the requirement of small residues in the hinge region17,38,39). Given these known coevolutionary pressures, it follows that there should be either highly conserved residues which are responsible for conferring this polymorphic behaviour, or a coevolutionary signal present in the sequences of functionally interacting regions within the serpin. As we were interested in the interactions between the residues of the RCL and residues beneath the RCL, we calculated conservation scores using a sequence alignment of 212 serpin sequences, and mapped them onto the structure of α1-AT (Fig. 3C). Residues facing the P1 and P1’ residues of the RCL are well conserved, compared to residues on strands s2C and s3C that face the RCL (under the residues N-terminal to P1). We were unable to identify any significant coevolutionary links between residues of the RCL and the region below it on sheet C, though this is most likely a reflection on the limited number of sequences used.
To further investigate the interactions between the RCL and the body of the serpin, we looked at the frustration networks within conserpin-AATRCL and α1-AT. Frustration analysis labels pairs of residues as ‘frustrated’ if their interaction is destabilising compared to other combinations of residues in the same location40; clusters of frustrated residues are often found near binding sites, suggestive of a stressed conformational state, or otherwise implicated in the function of the protein41. In α1-AT, the RCL is minimally frustrated against the body of the serpin, with only the P12-P9 region present in a patch of high frustration. In contrast, there is a more extensive network of frustration in conserpin-AATRCL, particularly between the RCL and the loop between s3A and s3C (Fig. S2). These distinct frustration patterns reflect the differences we observed in the electrostatics on top of the serpin body (Fig. 3), and suggest that the electrostatic compatibility between the body of the serpin and the RCL plays a key role in determining serpin functionality.
Having established clear differences in the surface electrostatics of the serpins, we next investigated possible consequences for engagement with proteases trypsin and HNE. Given contrasting inhibition of these two proteases we compared their electrostatic potential surfaces. The largest difference between the two proteases is found at the active site. Whereas both proteases feature an electronegative potential in the active site cleft, in trypsin it is more extensive, encompassing S2–S4 binding pockets and the surrounding residues (Fig. 4B,E). In contrast, the S3–S4 binding pockets and surrounding residues of HNE contains an adjacent large electropositive patch (Fig. 4E). To observe any electrostatic potential clashes during a hypothetical serpin−protease encounter complex, we modelled a conserpin-AATRCL: trypsin complex, and a conserpin-AATRCL: HNE complex, each with P1 M358329 in the protease active site (Fig. 4A,D), using the x-ray crystallography structure of a Michaelis complex as a starting model (PDB: 1K9O42). The electrostatic potential for each protease and serpin were calculated separately, eliminating the influence of one electrostatic potential onto the other.

Electrostatic compatibility between serpin and protease. (A) Electrostatic surfaces of a modeled complex between trypsin and conserpin-AATRCL, and (D) between HNE and conserpin-AATRCL. Associated complexes are separated into individual proteins by rotating each molecule by 90° around the horizontal axis in the plane of the paper (clockwise for the top molecules, anti-clockwise for the bottom molecules). (B) Electrostatic surface for the active site of trypsin and (E) HNE shows that trypsin has a more electronegative binding cleft than HNE. Comparing this to the electrostatic surface of (C, F idem.) conserpin-AATRCL suggests a greater electrostatic compatibility between trypsin, particularly the electropositive surface below the RCL. However, the electropositive surface of S3–S4 binding pocket in HNE suggests there may be a charge repulsion with the electropositive surface potential of conserpin-AATRCL at P6-P3.
The calculated electrostatic potential suggests trypsin has greater electrostatic compatibility with conserpin-AATRCL than HNE. This compatibility can be attributed to the large electropositive surface of the RCL and the body below the RCL. Compatibility will be essential for the formation and stability of a Michaelis serpin−protease complex, where there is contact between the binding pockets (S4−S1′) of the protease and P6−P1’ residues of the RCL. The formation and stability of a serpin−protease complex between conserpin-AATRCL and trypsin can occur with favourable interaction between trypsin’s electronegative S3–S4 pockets (Fig. 4B) and the electropositive potential of conserpin-AATRCL P6−P3 residues (Fig. 4C). Therefore, conserpin-AATRCL can inhibit trypsin. In comparison, the formation and stability of a complex may be hindered by the charge−charge repulsion between the electropositive S3–S4 binding pockets of HNE (Fig. 4D) and the electropositive surface of P6−P3 of the RCL (Fig. 4E). As a result, conserpin-AATRCL behaves as a substrate to HNE rather than as an inhibitor.
RCL dynamics are important for protease inhibition
Given the large conformational changes involved in serpin function43, and specifically the central role played by the RCL in protease engagement and subsequent insertion into the A-sheet, an investigation of the dynamics of the RCL of conserpin-AATRCL may provide some insight into its inhibitory properties. We therefore performed molecular dynamics (MD) simulations of conserpin-AATRCL and compared the results to those of α1-AT and conserpin simulations we performed previously31. Although we are unable to perform simulations for long enough to observe the RCL insertion into the A-sheet, MD is able to reveal the intrinsic dynamics of the RCL and specifically the lifetime of its interactions with the body of the serpin. After reaching equilibrium at around 150 ns, the root mean square deviation (RMSD) indicated that the simulations remained stable with no large conformational changes observed (Fig. S3). Given the importance of RCL conformation in facilitating the S→R transition following protease engagement, we analyzed the dynamics of the A-sheet and the RCL, and also the interactions between the RCL and the body of the serpin during the time course of the MD simulations. The central A-sheet contains two conserved regions, the shutter and breach, which are critical for the insertion of the RCL; mutations in these regions often render the serpin susceptible to misfolding and aggregation5.
Substitution of residues P7−P2’ of the RCL for the corresponding region of α1-AT did not serve to reduce the flexibility of the RCL region to the lower level observed in α1-AT simulations31. Instead, while the region of conserpin-AATRCL around residues 353324-362333 showed a reduced root mean square fluctuation (RMSF) (corresponding to less conformational variability), the region around residues 342314-352323 of the RCL showed an increased RMSF (Fig. 5D). This increase in flexibility is evident in comparing MD snapshots of the three systems, where the substantially increased flexibility of the lower RCL region of conserpin-AATRCL is clearly evident (Fig. 5A–C). This highly dynamic region encompasses the hinge region of the RCL, the first residues that insert into the A-sheet.

The dynamics of the RCL is important for inhibition. Snapshots of conformations of the RCL from the MD runs at 50 ns intervals overlaid on static structure for the rest of the molecule, showing that (A) conserpin prefers an extended-hinge RCL conformation, (B) α1-AT prefers a bent-hinge RCL conformation, and (C) conserpin-AATRCL occupies both of these conformations. the increased flexibility of the lower RCL region (residues 342314-352323) relative to both conserpin and α1-AT. (D) Root mean square fluctuation (RMSF) calculated for the RCL region from the molecular dynamics simulations shows that the conserpin-AATRCL (red) has lower flexibility than conserpin (black) in the 353324-362333 region but a higher flexibility in the 342314-352323 region than conserpin and α1-AT (blue) (α1-AT numbering), reflecting the structural differences between the two conformational clusters occupied by the RCL.
As the fate of the serpin as either a substrate or inhibitor is determined by the competition between rates of RCL insertion and the de-acylation of the protease16, it is likely that an increase in RCL loop dynamics would slow the rate of insertion. This would allow a protease with a fast catalytic mechanism (such as HNE), to escape inhibition, thereby pushing the serpin down the substrate pathway.
To further characterise the difference in the dynamics between the RCL of α1-AT and conserpin-AATRCL, we scrutinized the occupancy of salt bridges formed between the RCL and the body of the serpin over the course of the simulations. α1-AT contains 5 residues that can form salt bridges (1 aspartic acid, 1 lysine and 3 glutamic acids), while conserpin-AATRCL contains 4 residues (4 glutamic acids). Focusing on the salt bridges with >20% occupancy during the simulations, the occupancy difference of one salt bridge between the two simulations was most notable. The highly conserved salt bridge E342313-K290261 has a lower occupancy in conserpin-AATRCL than in α1-AT (41% compared to 91%). This salt bridge is the basis of the disease-causing Z variant of α1-AT, with removal of this interaction by the E342K mutation producing an aggregation-prone serpin39,44. The consequence of this decreased occupancy is an increase in the dynamics of E342313, increasing the dynamics in the hinge region, while leading to a decrease in the rate of RCL insertion, as previously observed45. Conserpin-AATRCL is capable of inhibiting trypsin as proteases differ in the rates of deacylation insertion, allowing for the de-acylation step of the HNE cleavage to occur, resulting in cleavage inactivation of conserpin-AATRCL and release of active HNE.
To understand the RCL conformations adopted by conserpin-AATRCL throughout the simulations, we performed principal component analysis on the conformations of the RCL backbone (between P17-P1’) over all simulations (α1-AT, conserpin and conserpin-AATRCL), followed by a clustering. This produced a total of 9 clusters, with RCL conformations within each cluster being structurally close but clearly distinguishable from others (Fig. S4). These analyses show that α1-AT’s RCL maintains a reasonably close set of conformations throughout the three independent simulations, while conserpin’s RCL explores a broad variety of conformations that are exclusive of those explored by α1-AT’s RCL (Fig. 6A). Conserpin-AATRCL’s RCL not only adopts conformations that overlap with those of the other serpins, but also explores conformations that were not seen in α1-AT and conserpin simulations.

RCL conformational cluster determination by principal component analysis. To describe the motion of the RCL across all simulations, principal component vectors were determined for all RCL backbone conformations. (A) The trajectories of each RCL (α1-AT: blue, conserpin: black, conserpin-AATRCL: red) are projected on the first 2 PC axes, and (B) these conformations were grouped into 9 clusters. For (C) conserpin, (D) conserpin-AATRCL and (E) α1-AT, representative RCL backbone conformations for the clusters explored by each serpin over the course of the MD simulations, are shown atop a serpin body (grey cartoon α1-AT (PDB: 3NE4)).
Conserpin’s RCL explored 5 different conformations (5 clusters), most of which have an extended conformation in which the hinge region (P12-P9) of the RCL is moved away from the breach region of β-sheet A (Fig. 6B). This preference for an extended RCL hinge in conserpin is surprising, as conserpin has an extended salt bridge network in the breach region in comparison to α1-AT31, which was hypothesised to stabilise conserpin’s native state.
For α1-AT, the RCL explores 2 similar conformations, with both conformations containing the hinge region primed for insertion between s3A and s5A. This expands on the RMSF analysis (Fig. 5D), where α1-AT’s RCL was seen to be relatively rigid over the course of the simulations (in comparison to conserpin and conserpin-AATRCL). The rigidity of α1-AT’s RCL suggests that there are interactions between the body and the RCL that reduce the dynamics of the loop, and possibly prime the hinge region between s3A and s5A strands.
The RCL of conserpin-AATRCL explores 4 conformations: one overlapping with a cluster seen in conserpin, one overlapping with a cluster seen in α1-AT, and 2 conformations unique to conserpin-AATRCL. All of these conformations (except the α1-AT-like one) include an extended hinge region away from β-sheet A. This could possibly be a consequence of the interactions between the residues on β-sheet C and the RCL, as stated previously26. Interestingly, one of the conformations include a slight helical turn from P10-P7 (similarly to an α1-AT / α1-antichymotrypsin chimera46), possibly responsible for pulling the hinge region away from β-sheet A. Importantly, one of conserpin-AATRCL’s RCL conformation is similar to α1-AT’s, where the hinge region is primed to insert into β-sheet A. The ability of conserpin-AATRCL to access this conformation may explain the increase in inhibitory activity against trypsin over conserpin, as the conserpin-AATRCL RCL could insert faster than conserpin from this pose. However, despite this primed hinge region conformation, conserpin-AATRCL primarily remains a substrate against HNE. It is possible that HNE could negatively impact on the conformation of the RCL upon encounter, or even prevent formation of a stable serpin:protease complex, a scenario in which HNE’s catalytic mechanism occurs more rapidly than trypsin, allowing for rapid cleavage of the RCL followed by substrate rather than inhibitor behaviour. A structural difference was also observed by calculating the phi-psi angles of the RCL for each serpin over the course of the simulations. Replacement of P7-P2’ of α1-AT onto conserpin has failed to reproduce the conformational pattern seen in α1-AT. Specifically, while the conformations adopted by conserpin-AATRCL in the region around residues 353324-362333 (Fig. S5, red) are more similar to those of α1-AT than conserpin (Fig. S5, blue and black, respectively), those in the region around residues 342314-352323 (Fig. S5) show a distinctly different set of conformations. Together these observations indicate that the conformational landscape sampled by the structure in and around the RCL region, including areas near the breach region, is important in the process of RCL insertion, and thus ultimately serpin inhibitory specificity.
Discussion
Conserpin shares high sequence identity to α1-AT (59%), is extremely stable, polymerisation-resistant and yields large quantities when expressed through recombinant techniques. It is therefore an attractive model system for investigating the folding, stability and function of serpins. In this study, to investigate the determinants of serpin specificity, we used a conserpin/α1-AT chimera, which we call conserpin-AATRCL, in which the residues in the RCL are replaced with those of α1-AT. The resulting hybrid retained the thermostability and polymerisation resistance of conserpin. However, despite containing the RCL sequence of α1-AT, which is thought to be a key determinant of inhibitory specificity, conserpin-AATRCL showed only minor improvement as an inhibitor of trypsin, in comparison to conserpin, and like conserpin behaved mostly as a substrate against HNE.
We attempted to rationalise the substrate behaviour of conserpin-AATRCL using a structural and molecular modelling/simulation approach. Although the x-ray crystal structure of conserpin-AATRCL revealed no significant differences with the parent molecule, we were able to provide insights into the failure to transfer specificity by analysing electrostatic differences and changes in the flexibility of RCL, hinge, breach and shutter regions with molecular dynamics simulations.
For a serpin to perform its inhibitory function, the serpin and protease must come into contact with each other. Reasoning that, like other protein−protein complexes47,48, cognate serpins and proteases must exhibit complementary electrostatic surfaces to ensure rapid, and high affinity association, we identified several differences between the electrostatic surface characteristics of α1-AT and conserpin-AATRCL that may contribute to their contrasting inhibitory properties. HNE contains a shallow active site that interacts with P6−P3’ residues of the RCL, therefore the electrostatic surface of this region must be complementary to ensure efficient binding to the P1 methionine residue27,49. In comparison to α1-AT, conserpin-AATRCL harbours several regions where poor charge complementarity may explain the diminished capacity to form a complex with HNE, and subsequently why it acts as a substrate rather than an inhibitor. One of these regions includes the electrostatic potential beneath the RCL. The role of electrostatics has been investigated for several serpins. For example, single-pair Förster resonance energy transfer (spFRET) studies of the inhibition of anionic rat and cationic bovine trypsin by α1-AT showed only partial translocation of anionic rat trypsin compared to full translocation of cationic bovine trypsin50,51. This indicates that the electrostatic potential between the protease and serpin are important for formation of a serpin:protease complex and protease inhibition. Similarly, for the serpin PAI-1, the Michaelis complex between tissue-type plasminogen activator (tPA) and PAI-1 was observed to have more complementary electrostatic interactions than the complex between urokinase-type plasminogen activator (uPA) and PAI-1. This was used to explain the difference in second-order inhibitory rate constants between the two proteases: tPA is inhibited at a faster rate (2.6 × 107 M−1 s−1) compared to uPA (4.8 × 106 M−1 s−1)52,53. Furthermore, an arginine to glutamic acid substitution produced a ‘serpin-resistant’ tPA variant, where the glutamic acid produced a repulsion to PAI-1, leading to a failure to inhibit this variant. This tPA variant was only inhibited through creating a complementary PAI-1 with the opposite glutamic acid to arginine mutation54,55, further emphasising the importance of surface potential in the formation of a stable serpin:protease complex. Any possible repulsive interactions may destabilize a serpin:protease complex and therefore prevent inhibition.
Dynamics in the RCL is important for its insertion into β-sheet A during protease inhibition. We therefore investigated the difference in RCL dynamics between conserpin-AATRCL and α1-AT using molecular dynamics simulations. Despite P7-P2’ of conserpin-AATRCL being identical to the corresponding region in α1-AT, the overall flexibility of the RCL region as a whole was not reduced to the level of α1-AT, while the hinge region of the RCL, which inserts into β-sheet A first during insertion, exhibited higher flexibility in conserpin-AATRCL compared to α1-AT. A plausible explanation for this is the additional residue at P2’. Conserpin was designed without an isoleucine at P2’, producing an RCL length that fits onto the serpin body. The addition of P2’ isoleucine in conserpin-AATRCL may force the RCL to adopt a non-ideal conformation, possibly increasing the dynamics of the hinge region.
The conformation of the RCL is likely highly tailored to the particular inhibitory specificity of each serpin. α1-AT, a potent inhibitor of HNE, has an RCL that is in a primed position for insertion into β-sheet A. That is, the hinge region is poised between strands 3A and 5A, allowing for rapid insertion during HNE inhibition. Conserpin and conserpin-AATRCL contain RCL conformations that are extended, with the hinge region away from β-sheet A, likely reducing the rate at which the RCL can insert. One conformation that conserpin-AATRCL explores contains a primed hinge region, which possibly explains the increase in its inhibitory activity against trypsin (compared to conserpin), but is not enough to produce inhibition against HNE. Furthermore, the breach region of α1-AT ‘loosens’ and opens over the course of the simulations31, while the breach region in conserpin and conserpin-AATRCL remains rigid due to the extended salt bridge network. Therefore, it is possible that α1-AT inhibits HNE at a rapid rate due to the primed position of the hinge region and opening of the breach region, allowing inhibition before HNE’s de-acylation step of cleavage. Our observation that the RCL of conserpin-AATRCL sampled this primed hinge conformation out of 4 possible conformations, and the relatively rigid nature of its breach region, suggests that the initial steps of RCL insertion into A-sheet are slower than the de-acylation step of HNE’s cleavage.
Previous studies that have attempted to convert the specificity of ACT to that of α1-AT by swapping RCL residues have been generally unsuccessful, as the chimeras had a greater SI and slower inhibitory rate compared to α1-AT. This suggests that other factors may play important roles, including interactions between the RCL and the body of the serpin, and the structure of the chimeric RCL15,26,27,46. Engineering of ACT/α1-AT chimeras show that HNE’s proteolytic mechanism occurs on a shorter timescale in comparison to ACT’s catalytic mechanism15. It is also known that an increase in the dynamics of the RCL can affect the serpin’s ability to inhibit a protease. Notably, loss of a salt bridge in the breach region in α1-AT Z variant increases RCL dynamics and subsequently leads to an SI increase (from 1.0 to 1.8) and decrease in rate of inhibition (from 6.9 to 2.3 × 106 M−1 s−1)45,56,57,58. This implies that the rate of RCL insertion occurs slower than the de-acylation step of HNE’s catalytic mechanism, producing a substrate rather than an inhibitor of HNE. It is likely that RCL-protease interactions will vary for each protease, influencing the dynamics of the RCL29 and the conformational change needed for RCL insertion59. With the use of fluorescent labels, it was observed that the two protease targets of plasminogen activator inhibitor-1 (PAI-1), tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA), rests differently on the P1−P1’ bond and change the dynamics of the RCL when bound. tPA affects the C-terminus of the RCL through exosite interactions, while retaining dynamics observed with free PAI-1. In contrast, uPA affects the N-terminus with different exosite interactions, restricting the dynamics and immobilising the RCL. This difference in RCL dynamics also contributes to the difference in the rates the proteases are inhibited by PAI-129. Taken together, the dynamics of the RCL is critical for the rate of insertion during protease inhibition. Fast insertion favours protease inhibition while slow insertion forces the serpin to undergo the substrate pathway.
Along with the possibility of electrostatic repulsion and increased RCL dynamics, the failure to transfer specificity onto conserpin-AATRCL could be a consequence of the delicate balance between stability and function. Serpins use the metastable conformation to undergo the large conformational change necessary for its inhibitory function. Increasing the stability of this metastable state may decrease the dynamics and plasticity required to undergo the S → R transition during inhibition of a target protease. For example, increasing the stability of α1-AT more than 13 kcal mol−1 than the wild type α1-AT compromises its inhibitory activity60. For conserpin, the very high stability, although still functional, can be attributed to certain key regions important for the serpin’s inhibitory mechanism and S → R transition31. Structural plasticity is required in the breach region, as this region is important in controlling the insertion of the RCL and conformational change to allow for protease inhibition. The extensive salt-bridge network in the breach region in conserpin and conserpin-AATRCL increases rigidity and slows the opening of β-sheet A between strands s3A and s5A. The rigidity of the breach, along with displacement of conserpin and conserpin-AATRCL hinge region away from β-sheet A, may explain the reduced inhibitory activity of conserpin towards trypsin, and the failure of conserpin-AATRCL to inhibit HNE. Furthermore, helix F, which packs tightly against the A-sheet, may act as a barrier to RCL insertion via A-sheet opening, and must partially unfold to allow rapid RCL insertion61,62,63. Mutations on the helix F/A-sheet interface of α1-AT can relieve this tight packing, increasing the stability but also decreasing activity. Therefore, the tight packing between helix F and A-sheet contributes to the metastability and that is relieved in the S → R transition. In conserpin, this interface is tightly packed, but not to the extent of α1-AT. As a result, the tight packing at this interface may not have the strain observed in α1-AT, slowing the partial unfolding of helix-F to allow for rapid RCL insertion.
In conclusion, we utilized a serpin chimera to investigate the rules that govern serpin specificity, by studying the effect of replacing the RCL of conserpin, a model synthetic serpin, with the corresponding sequence from α1-AT. Despite possessing the RCL sequence of α1-AT, specificity against trypsin or HNE was not restored to that of α1-AT. Crystal structural analysis and molecular dynamics simulations indicate that, although the RCL sequence may partially dictate specificity, electrostatic surface potential coupled with dynamics in and around the RCL likely play an important role. Although beyond the scope of the current study, systematic mutational studies on conserpin-AATRCL that alter its electrostatic complementarity with HNE will ultimately allow our hypotheses to be tested. The dynamics of the RCL appears to govern the rate of insertion during protease inhibition, dictating whether it behaves as an inhibitor or a substrate. The unusual mechanism of serpin action also requires a delicate balance between stability, dynamics and function64,65. Engineering serpin specificity is therefore substantially more complex than solely manipulating the RCL sequence, and although may be guided by the general principles discussed in this work, each serpin will most likely present unique challenges. Notwithstanding this, further characterisation of the role of dynamics will be required to advance our understanding of how serpins perform their exquisite inhibitory functions.
Materials and Methods
Design of conserpin-AATRCL
The design of conserpin-AATRCL was based of the RCL sequence of α1-AT. Residues P7-P2’ of the α1-AT RCL were mutated onto the original conserpin molecule to provide specificity against trypsin and neutrophil elastase. The residue numbering adheres to that adopted as previous31: Q105α1-AT and corresponding conserpin-AATRCL residue R79conserpin-AATRCL is written as Q105R79.
Expression constructs
The plasmid encoding conserpin-AATRCL was generated using ligation-independent cloning with the pLIC-HIS vector66 using standard protocols, adding an N-terminal 6His-tag to conserpin-AATRCL; this construct was transformed into BL21(DE3) pLysS E. coli.
Protein expression and purification
Protein was expressed 2xYT media and induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) at an OD600 of 1. Expression was continued for 3 hours before cells were harvested and lysed in 10 mM imidazole, 50 mM NaH2PO4, 300 mM NaCl, pH 8.0. Following centrifugation, batch bound to nickel-NTA loose resin (Qiagen) and washed with 50 mL of 20 mM imidazole, 50 mM NaH2PO4, 300 mM NaCl, pH 8.0. Any conserpin-AATRCL bound to the nickel-NTA resin was eluted with 250 mM imidazole, 50 mM NaH2PO4, 300 mM NaCl, pH 8.0, into 5 mL fractions. Fractions containing conserpin-AATRCL were loaded into a Superdex 200 16/60 column for further purification and eluted with 50 mM tris-HCl, 150 mM NaCl pH 8.0. The N-terminal His-tag remained attached to conserpin-AATRCL.
Characterisation of inhibitory properties
The stoichiometry of inhibition against bovine trypsin (Sigma-Aldrich) was performe similarly as described31,36. Briefly, various concentrations of conserpin-AATRCL (0−200 nM in 25 nM increments) was incubated with a constant trypsin (105 nM) concentration at 37 °C for 30 min in 50 mM tris-HCl, 150 mM NaCl, 0.2% v/v PEG 8000 pH 8.0. The residual trypsin activity was measured at 405 nm using the substrate Na-benzoyl-L-arginine 4-nitroanilide hydrochloride (Sigma-Aldrich).
To test for activity after refolding, conserpin-AATRCL was unfolded in 6 M guanidine hydrochloride (GndHCl) 50 mM tris-HCl, 150 mM NaCl pH 8.0 for 2 hours before refolding via dilution for another 2 hours, so the final concentration of guanidine hydrochloride was 0.2 M. Any aggregate was pelleted by centrifugation and the sample dialysed against the same buffer to remove any remaining GndHCl. The SI assay against trypsin was performed as stated above (constant trypsin concentration of 210 nM and varying conserpin-AATRCL concentrations from 0−450 nM in 50 nM increments).
To observe an SDS-stable serpin: protease complex, different ratios of serpin were incubated with protease for 30 minutes at 37 °C. Reducing SDS sample buffer was added to each sample and quenched on ice to stop any further reaction. Samples were loaded onto a 10% SDS-PAGE.
Circular dichroism scans and thermal denaturation
Circular dichroism (CD) measurements were performed on a Jasco J-815 CD spectrometer at a protein concentration of 0.2 mg/mL with PBS using a quartz cell with a path-length of 0.1 cm. Far-UV scans were performed at 190−250 nm. For thermal denaturation, a heating rate of 1 °C/min from 35 °C to 95 °C was used, with the change in signal measured at 222 nm. For samples containing 2 M GdnHCl, refolding was measured directly after the thermal melt by holding the temperature at 95 °C for 1 min before the temperature was decreased to 35 °C at the same rate. The midpoint of transition (Tm) was obtained by fitting the data with a Boltzmann sigmoidal curve in accordance with the method described31 for both forward and reverse thermal denaturation experiments.
Crystallization, X-ray data collection, structure determination and refinement
Crystals of conserpin-AATRCL were obtained using hanging drop vapour diffusion, with 1:1 (v/v) ratio of protein to mother liquor (1 μL of conserpin-AATRCL mixed with 1 μL of mother liquor. The protein was concentrated to 10 mg/mL and crystals appeared in 0.2 M magnesium chloride, 0.1 mM Bis-Tris and 20% PEG 3350, pH 6.5 after 5 days.
Diffraction data was collected on the MX2 beamline at the Australian Synchrotron. The diffraction data was processed with iMOSFLM67 to 2.48 Å, followed by scaling with SCALA68 in the CCP4 suite69. The structure was determined by molecular replacement (MR) with Phaser70 using the conserpin structure (native state) as a search probe (PDB 5CDX)31. The model was built and refined using PHENIX71 and Coot72.
Computational resources Atomistic MD simulations were performed on Multi-modal Australian ScienceS Imaging and Visualisation Environment (MASSIVE), and in-house hardware (NVIDIA TITAN X Pascal GPU).
Atomic coordinates, modelling and graphics
The RCL was modelled onto the x-ray crystal structure using MODELLER73. In MD simulations, atomic coordinates were obtained from the following PDB entry: 3NE437. α1-AT and conserpin MD simulations used for the analysis used our previously reported data31. The residue numbering remained as determined by crystal structure, that is, the glutamine from the TEV cleavage tag remained as residue -1. Structural representations were produced using PyMOL version 2.0.474 and VMD 1.9.475. Trajectory manipulation and analysis was performed using MDTraj76 and VMD 1.9.475. Electrostatic calculations were performed with the APBS plugin77,78 on PyMOL. Serpin:protease complexes were modelled based on the X-ray crystal structure of a serpin:protease Michaelis complex (Manduca sexta serpin 1B with rat trypsin (S195A), PDB: 1K9O)42.
Molecular dynamics (MD) systems setup and simulation
Each protein, with protonation states appropriate for pH 7.0 as determined by PROPKA79,80, was placed in a rectangular box with a border of at least 10 Å, explicitly solvated with TIP3P water81, sodium counter-ions added, and parameterized using the AMBER ff99SB all-atom force field82,83,84. After an energy minimization stage consisting of at least 10,000 steps, an equilibration protocol was followed in which harmonic positional restraints of 10 kcal Å2 mol−1 were applied to the protein backbone atoms. The temperature was incrementally increased while keeping volume constant from 0 K to 300 K over the course of 0.5 ns, with Langevin temperature coupling relaxation times of 0.5 ps. After the target temperature was reached, pressure was equilibrated to 1 atm over a further 0.5 ps using the Berendsen algorithm85. Following equilibration, production runs were performed in the NPT ensemble using periodic boundary conditions and a time step of 2 fs. Temperature was maintained at 300 K using the Langevin thermostat with a collision frequency of 2 ps, and electrostatic interactions computed using an 8 Å cutoff radius and the Particle Mesh Ewald method86. Three independent replicates of each system were simulated for 500 ns each using Amber 1487. The three independent replicates for each system were concatenated, and RMSD, RMSF, and phi and psi angles computed over 500 ps timesteps using VMD 1.9.475.
Sequence methods
In calculating construct sequence identities, construct sequences were aligned using MUSCLE88,89 v3.8.1551. The 6xHIS-TEV-SacII N-terminal peptide was removed from the alignment so as not to inflate alignment statistics. Percentage identities were calculated as %id = 100% × number of identity columns/length of aligned region (including gaps).
Mapping of sequence conservation on structure α1-AT90 was performed using the Consurf 2016 server91 using the previously designed alignment of 212 serpins39. Sequence coevolution analysis was performed using the OMES χ2 residue independence test92, as well as the SCA93 and ELSC94 perturbation-based residue covariance methods.
RCL principal component analysis & clustering
From the nine trajectories described above, trajectories of the 72 atoms describing the backbone (N, CA, C, O) from P17 (E342)−P1’ (S359) were extracted using MDTraj95. These trajectories were concatenated together into a 8993-frame trajectory, and Scikit-learn95 was used to calculate eigenvectors describing 216 principal component vectors. The top three PCA vectors describe 35.64%, 16.67%, and 11.72% respectively of the variance across all conformations in the concatenated trajectory. The nine trajectories were then transformed into this PCA space, and plotted using matplotlib96.
The concatenated trajectory, as expressed in PCA coordinates, was clustered using the HDBSCAN algorithm97,98, using default parameters, except a minimum cluster size of 1% of the total trajectory (90 frames).
Frustration calculation
Local frustration analysis of the modelled serpin: protease complexes was conducted with the Frustratometer2 web server99. Essentially, the energetic frustration is obtained by the comparison of the native state interactions to a set of generated “decoy” states where the identities of each residue are mutated. The constant k used to model the electrostatic strength of the system was set to its default value (4.15). A contact is defined as “minimally frustrated” or “highly frustrated” upon comparison of its frustration energy with values obtained from the decoy states.
Accession Numbers
The coordinates and structure factors have been deposited in the Protein Data Bank under accession code 6EE5.
Data Availability
All data is available upon request from the corresponding author (AMB).
References
Huntington, J. A., Read, R. J. & Carrell, R. W. Structure of a serpin-protease complex shows inhibition by deformation. Nature 407, 923–926 (2000).
Law, R. H. P. et al. An overview of the serpin superfamily. Genome Biol. 7, 216 (2006).
Elliott, P. R., Lomas, D. A., Carrell, R. W. & Abrahams, J. P. Inhibitory conformation of the reactive loop of alpha 1-antitrypsin. Nat. Struct. Biol. 3, 676–681 (1996).
Stratikos, E. & Gettins, P. G. W. Formation of the covalent serpin-proteinase complex involves translocation of the proteinase by more than 70 Å and full insertion of the reactive center loop into β-sheet A. Proc. Natl. Acad. Sci. 96, 4808–4813 (1999).
Whisstock, J. C., Skinner, R., Carrell, R. W. & Lesk, A. M. Conformational changes in serpins: I. The native and cleaved conformations of alpha1-antitrypsin. J. Mol. Biol. 296, 685–699 (2000).
Tew, D. J. & Bottomley, S. P. Probing the equilibrium denaturation of the serpin alpha1-antitrypsin with single tryptophan mutants; evidence for structure in the urea unfolded state. J. Mol. Biol. 313, 1161–1169 (2001).
Gettins, P. G. W. Serpin structure, mechanism, and function. Chem. Rev. 102, 4751–804 (2002).
Krishnan, B. & Gierasch, L. M. Dynamic local unfolding in the serpin α1 antitrypsin provides a mechanism for loop insertion and polymerization. Nat. Struct. Mol. Biol. 18, 222–6 (2011).
Tsutsui, Y., Dela Cruz, R. & Wintrode, P. L. Folding mechanism of the metastable serpin α1-antitrypsin. Proc. Natl. Acad. Sci. USA 109, 4467–72 (2012).
Cabrita, L. D. & Bottomley, S. P. How do proteins avoid becoming too stable? Biophysical studies into metastable proteins. Eur. Biophys. J. 33, 83–88 (2004).
James, E. L. & Bottomley, S. P. The mechanism of alpha1-antitrypsin polymerization probed by fluorescence spectroscopy. Arch. Biochem. Biophys. 356, 296–300 (1998).
Dupont, D. M. et al. Biochemical properties of plasminogen activator inhibitor-1. Front. Biosci. Landmark Ed. 14, 1337–61 (2009).
Mushunje, A., Evans, G., Brennan, S. O., Carrell, R. W. & Zhou, A. Latent antithrombin and its detection, formation and turnover in the circulation. J. Thromb. Haemost. 2, 2170–2177 (2004).
Padrines, M., Schneider-Pozzer, M. & Bieth, J. G. Inhibition of neutrophil elastase by alpha1-proteinase inhibitor oxidized by activated neutrophils. Am. Rev. Respir. Dis. 139, 783–90 (1989).
Rubin, H. et al. Conversion of alpha1-antichymotrypsin into a human neutrophil elastase inhibitor: demonstration of variants with different association rate constants, stoichiometries of inhibition, and complex stabilities. Biochemistry 33, 7627–7633 (1994).
Lawrence, D. A. et al. Partitioning of serpin-proteinase reactions between stable inhibition and substrate cleavage is regulated by the rate of serpin reactive center loop insertion into β-sheet A. J. Biol. Chem. 275, 5839–5844 (2000).
Hopkins, P. C. R., Carrell, R. W. & Stone, S. R. Effects of mutations in the hinge region of serpins. Biochemistry 32, 7650–7657 (1993).
Rau, J. C., Beaulieu, L. M., Huntington, J. A. & Church, F. C. Serpins in thrombosis, hemostasis and fibrinolysis. J. Thromb. Haemost. 5(Suppl 1), 102–15 (2007).
Polderdijk, S. G. I. et al. Design and characterization of an APC-specific serpin for the treatment of hemophilia. Blood 129, 105–113 (2017).
Owen, M. C., Brennan, S. O., Lewis, J. H. & Carrell, R. W. Mutation of Antitrypsin to Antithrombin. N. Engl. J. Med. 309, 694–698 (1983).
Ehrlich, H. J. et al. Alteration of serpin specificity by a protein cofactor: Vitronectin endows plasminogen activator inhibitor 1 with thrombin inhibitory properties. J. Biol. Chem. 265, 13029–13035 (1990).
Lawrence, Da, Strandberg, L., Ericson, J. & Ny, T. Structure-Function Inhibitor Type 1 Studies of the SERPIN Plasminogen Activator. J. Biol. Chem. 265, 20293–20301 (1990).
Patston, P. A. et al. Reactivity of α1-antitrypsin mutants against proteolytic enzymes of the kallikrein-kinin, complement, and fibrinolytic systems. J. Biol. Chem. 265, 10786–10791 (1990).
Hopkins, P. C. R., Crowther, D. C., Carrell, R. W. & Stone, S. R. Development of a novel recombinant serpin with potential antithrombotic properties. J. Biol. Chem. 270, 11866–11871 (1995).
Chaillan-Huntington, C. E., Gettins, P. G. W., Huntington, J. A. & Patston, P. A. The P6-P2 region of serpins is critical for proteinase inhibition and complex stability. Biochemistry 36, 9562–9570 (1997).
Djie, M. Z., Stone, S. R. & Le Bonniec, B. F. Intrinsic specificity of the reactive site loop of α1-antitrypsin, α1-antichymotrypsin, antithrombin III, and protease nexin I. J. Biol. Chem. 272, 16268–16273 (1997).
Plotnick, M. I., Schechter, N. M., Wang, Z. M., Liu, X. & Rubin, H. Role of the P6-P3’ region of the serpin reactive loop in the formation and breakdown of the inhibitory complex. Biochemistry 36, 14601–14608 (1997).
Whisstock, J. C. et al. Serpins flex their muscle: II. Structural insights into target peptidase recognition, polymerization, and transport functions. J. Biol. Chem. 285, 24307–24312 (2010).
Qureshi, T., Goswami, S., McClintock, C. S., Ramsey, M. T. & Peterson, C. B. Distinct encounter complexes of PAI-1 with plasminogen activators and vitronectin revealed by changes in the conformation and dynamics of the reactive center loop. Protein Sci. 25, 499–510 (2016).
Gettins, P. G. W. & Olson, S. T. Exosite determinants of serpin specificity. J. Biol. Chem. 284, 20441–20445 (2009).
Porebski, B. T. et al. Smoothing a rugged protein folding landscape by sequence-based redesign. Sci. Rep. 6, 33958 (2016).
Yang, L., Irving, J. A., Dai, W., Aguilar, M. & Bottomley, S. P. Probing the folding pathway of a consensus serpin using single tryptophan mutants. Sci. Rep. 1–15, https://doi.org/10.1038/s41598-018-19567-9 (2018).
Zhou, A. et al. Polymerization of Plasminogen Activator Inhibitor-1. J. Biol. Chem. 276, 9115–9122 (2001).
Dafforn, T. R., Mahadeva, R., Elliott, P. R., Sivasothy, P. & Lomas, D. A. A Kinetic Mechanism for the Polymerization of α1-Antitrypsin. J. Biol. Chem. 274, 9548–9555 (1999).
Belorgey, D., Hägglöf, P., Onda, M. & Lomas, D. A. pH-dependent stability of neuroserpin is mediated by histidines 119 and 138; Implications for the control of β-sheet a and polymerization. Protein Sci. 19, 220–228 (2010).
Horvath, A. J., Lu, B. G. C., Pike, R. N. & Bottomley, S. P. Methods to measure the kinetics of protease inhibition by serpins. Methods in Enzymology. 501, 223–235 (2011).
Patschull, A. O. M. et al. Therapeutic target-site variability in α1-antitrypsin characterized at high resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 1492–1497 (2011).
Buck, M. J. & Atchley, W. R. Networks of Coevolving Sites in Structural and Functional Domains of Serpin Proteins. Mol. Biol. Evol. 22, 1627–1634 (2005).
Irving, J. A., Pike, R. N., Lesk, A. M. & Whisstock, J. C. Phylogeny of the serpin superfamily: Implications of patterns of amino acid conservation for structure and function. Genome Res. 10, 1845–1864 (2000).
Ferreiro, D. U., Hegler, J. A., Komives, E. A. & Wolynes, P. G. Localizing frustration in native proteins and protein assemblies. Proc. Natl. Acad. Sci. USA 104, 19819–24 (2007).
Jenik, M. et al. Protein frustratometer: a tool to localize energetic frustration in protein molecules. Nucleic Acids Res. 40, W348–W351 (2012).
Ye, S. et al. The structure of a Michaelis serpin-protease complex. Nat. Struct. Biol. 8, 979–983 (2001).
Kass, I., Reboul, C. F. & Buckle, A. M. Computational methods for studying serpin conformational change and structural plasticity. Methods Enzymol. 501, 295–323 (2011).
Lomas, D. A., Evans, D. L., Finch, J. T. & Carrell, R. W. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 357, 605–7 (1992).
Kass, I., Knaupp, A. S., Bottomley, S. P. & Buckle, A. M. Conformational properties of the disease-causing Z variant of α1-antitrypsin revealed by theory and experiment. Biophys. J. 102, 2856–65 (2012).
Wei, A., Rubin, H., Cooperman, B. S. & Christianson, D. W. Crystal structure of an uncleaved serpin reveals the conformation of an inhibitory reactive loop. Nat. Struct. Mol. Biol. 1, 251–258 (1994).
Schreiber, G. & Fersht, A. R. Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 3, 427–431 (1996).
Schreiber, G. & Fersht, A. R. Interaction of Barnase with Its Polypeptide Inhibitor Barstar Studied by Protein Engineering. Biochemistry 32, 5145–5150 (1993).
Plotnick, M. I., Rubin, H. & Schechter, N. M. The Effects of Reactive Site Location on the Inhibitory Properties of the Serpin α1 -Antichymotrypsin. J. Biol. Chem. 277, 29927–29935 (2002).
Liu, L., Mushero, N., Hedstrom, L. & Gershenson, A. Short-lived protease serpin complexes: partial disruption of the rat trypsin active site. Protein Sci. 16, 2403–2411 (2007).
Liu, L., Mushero, N., Hedstrom, L. & Gershenson, A. Conformational distributions of protease-serpin complexes: A partially translocated complex. Biochemistry 45, 10865–10872 (2006).
Gong, L. et al. Crystal structure of the Michaelis complex between tissue-type plasminogen activator and plasminogen activators inhibitor-1. J. Biol. Chem. 290, 25795–25804 (2015).
Lin, Z. et al. Structural basis for recognition of urokinase-type plasminogen activator by plasminogen activator inhibitor-1. J. Biol. Chem. 286, 7027–32 (2011).
Madison, E. L. et al. Amino acid residues that affect interaction of tissue-type plasminogen activator with plasminogen activator inhibitor 1. Proc. Natl. Acad. Sci. USA 87, 3530–3 (1990).
Madison, E. L., Goldsmith, E. J., Gerard, R. D., Gething, M. J. & Sambrook, J. F. Serpin-resistant mutants of human tissue-type plasminogen activator. Nature 339, 721–724 (1989).
Hughes, V. A., Meklemburg, R., Bottomley, S. P. & Wintrode, P. L. The Z mutation alters the global structural dynamics of α1-antitrypsin. PLoS One 9, e102617 (2014).
Levina, V. et al. Expression, purification and characterization of recombinant Z α1-Antitrypsin-The most common cause of α1-Antitrypsin deficiency. Protein Expr. Purif. 68, 226–232 (2009).
Huang, X. et al. Molecular mechanism of Z α1-antitrypsin deficiency. J. Biol. Chem. 291, 15674–15686 (2016).
Lee, K. N., Im, H., Kang, S. W. & Yu, M. H. Characterization of a human alpha1-antitrypsin variant that is as stable as ovalbumin. J. Biol. Chem. 273, 2509–2516 (1998).
Seo, E. J., Lee, C. & Yu, M. H. Concerted regulation of inhibitory activity of α1-antitrypsin by the native strain distributed throughout the molecule. J. Biol. Chem. 277, 14216–14220 (2002).
Cabrita, L. D., Whisstock, J. C. & Bottomley, S. P. Probing the role of the F-helix in serpin stability through a single tryptophan substitution. Biochemistry 41, 4575–81 (2002).
Cabrita, L. D., Dai, W. & Bottomley, S. P. Different conformational changes within the F-helix occur during serpin folding, polymerization, and proteinase inhibition. Biochemistry 43, 9834–9839 (2004).
Tsutsui, Y., Liu, L., Gershenson, A. & Wintrode, P. L. The Conformational Dynamics of a Metastable Serpin Studied by Hydrogen Exchange and Mass Spectrometry. Biochemistry 45, 6561–6569 (2006).
Fulton, K. F. et al. The high resolution crystal structure of a native thermostable serpin reveals the complex mechanism underpinning the stressed to relaxed transition. J. Biol. Chem. 280, 8435–8442 (2005).
Zhang, Q. et al. The N terminus of the serpin, tengpin, functions to trap the metastable native state. EMBO Rep. 8, 658–63 (2007).
Cabrita, L. D., Dai, W. & Bottomley, S. P. A family of E. coli expression vectors for laboratory scale and high throughput soluble protein production. BMC Biotechnol. 6, 12 (2006).
Battye, T. G. G., Johnson, O., Powell, H. R. & Leslie, A. G. W. iMOSFLM: a new graphical interface for diffraction- image processing with MOSFLM research papers. Acta Crystallogr. D Biol. Crystallogr 67, 271–281 (2011).
Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 62, 72–82 (2006).
Winn, M. D., Ballard, C. C., Cowtan, K. D. & Dodson, E. J. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 235–242 (2011).
McCoy, A. J. Phaser crystallographic software. J Appl Crystallogr 40, 658–674 (2007).
Adams, P. D. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P. & Cowtan, K. COOT: Model-Building Tools for Molecular Graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 (2004).
Eswar, N. et al. Comparative protein structure modeling using Modeller. In Current Protocols in Bioinformatics 5–6, https://doi.org/10.1002/0471250953.bi0506s15 (2002).
Schrodinger, L. The Pymol Molecular Graphics System, Version 2.0.4.
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Mcgibbon, R. T. et al. MDTraj: a modern, open library for the analysis of molecular dynamics trajectories MDTraj: a modern, open library for the analysis of molecular dynamics trajectories. Biorxiv.Org 109, 0–2 (2014).
Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. 98, 10037–10041 (2001).
Jurrus, E. et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018).
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 (2004).
Søndergaard, C. R., Olsson, M. H. M. & Rostkowski Michałand Jensen, J. H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J Chem Theory Comput 7, 2284–2295 (2011).
Jorgensen, W. L. et al. Comparison of simple potential functions for simulating liquid water Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 926, 926–935 (2001).
Suk Joung, I. & Cheatham, T. E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 112, 9020–9041 (2008).
Maier, J. A. et al. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Li, P., Roberts, B. P., Chakravorty, D. K. & Merz, K. M. Rational design of particle mesh ewald compatible lennard-jones parameters for +2 metal cations in explicit solvent. J. Chem. Theory Comput. 9, 2733–2748 (2013).
Berendsen, H. J. C., Postma, J. P. M., Van Gunsteren, W. F., Dinola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Case, D. A. et. al. Amber 14. (University of California, San Francisco, 2014).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Landau, M. et al. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, 299–302 (2005).
Ashkenazy, H. et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016).
Kass, I. & Horovitz, A. Mapping pathways of allosteric communication in GroEL by analysis of correlated mutations. Proteins Struct. Funct. Genet. 48, 611–617 (2002).
Lockless, S. W. & Ranganathan, R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286, 295–9 (1999).
Dekker, J. P., Fodor, A., Aldrich, R. W. & Gary, Y. A perturbation-based method for calculating explicit likelihood of evolutionary co-variance in multiple sequence alignments. Bioinformatics 20, 1565–1572 (2004).
McGibbon, R. T. et al. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 109, 1528–32 (2015).
Hunter, J. D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 9, 99–104 (2007).
Campello, R. J. G. B., Moulavi, D. & Sander, J. Density-Based Clustering Based on Hierarchical Density Estimates. in. Antimicrobial agents and chemotherapy 58, 160–172 (2013).
McInnes, L., Healy, J. & Astels, S. hdbscan: Hierarchical density based clustering. J. Open Source Softw. 2, 11–12 (2017).
Parra, R. G. et al. Protein Frustratometer 2: a tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic Acids Res. 44, W356–W360 (2016).
Acknowledgements
BTP is a Medical Research Council Career Development Fellow. SM acknowledges fellowship support from the Australian Research Council (FT100100960). We thank the Australian Synchrotron for beam-time and technical assistance. This work was supported by the Multi-modal Australian ScienceS Imaging and Visualisation Environment (MASSIVE) (www.massive.org.au). We acknowledge the Monash Protein Production Unit and Monash Macromolecular Crystallization Facility. The coordinates and structure factors have been deposited in the Protein Data Bank under accession code 6EE5.
Author information
Authors and Affiliations
Contributions
E.M.M. and A.M.B. designed the study. E.M.M. performed the protein expression, purification, C.D. thermal melt and assay experiments with assistance from B.T.P. and D.E.H. E.M.M., S.M. and A.M.B. performed the crystallography, with assistance from B.T.P. J.F. performed molecular dynamics simulations. J.F. and B.T.R. analyzed simulations, with assistance from I.K. M.G.S.C. performed frustration analysis. E.M.M. and B.T.R. generated figures. E.M.M., B.T.R., S.M. and A.M.B. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marijanovic, E.M., Fodor, J., Riley, B.T. et al. Reactive centre loop dynamics and serpin specificity. Sci Rep 9, 3870 (2019). https://doi.org/10.1038/s41598-019-40432-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40432-w
This article is cited by
-
Enoxaparin augments alpha-1-antitrypsin inhibition of TMPRSS2, a promising drug combination against COVID-19
Scientific Reports (2022)
-
Deep mutational scanning of the plasminogen activator inhibitor-1 functional landscape
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
