
Abstract
Spotted fever group (SFG) rickettsiae are obligate intracellular Gram-negative bacteria mainly associated with ticks. In Japan, several hundred cases of Japanese spotted fever, caused by Rickettsia japonica, are reported annually. Other Rickettsia species are also known to exist in ixodid ticks; however, their phylogenetic position and pathogenic potential are poorly understood. We conducted a nationwide cross-sectional survey on questing ticks to understand the overall diversity of SFG rickettsiae in Japan. Out of 2,189 individuals (19 tick species in 4 genera), 373 (17.0%) samples were positive for Rickettsia spp. as ascertained by real-time PCR amplification of the citrate synthase gene (gltA). Conventional PCR and sequencing analyses of gltA indicated the presence of 15 different genotypes of SFG rickettsiae. Based on the analysis of five additional genes, we characterised five Rickettsia species; R. asiatica, R. helvetica, R. monacensis (formerly reported as Rickettsia sp. In56 in Japan), R. tamurae, and Candidatus R. tarasevichiae and several unclassified SFG rickettsiae. We also found a strong association between rickettsial genotypes and their host tick species, while there was little association between rickettsial genotypes and their geographical origins. These observations suggested that most of the SFG rickettsiae have a limited host range and are maintained in certain tick species in the natural environment.
Similar content being viewed by others

Introduction
Rickettsiae are obligate intracellular Gram-negative bacteria that belong to the order Rickettsiales in the class Alphaproteobacteria1. The members of the genus Rickettsia are divided into four main groups: the spotted fever group (SFG), typhus group (TG), transitional group (TRG), and ancestral group (AG)2. SFG and AG rickettsiae are mainly associated with ticks, while TG and TRG rickettsiae are associated with other arthropods such as lice, fleas, and mites. TG is composed of Rickettsia typhi and R. prowazekii, while TRG is composed of R. akari, R. australis, and R. felis. Among the tick-borne rickettsiae, AG includes R. bellii and R. canadensis. More than 25 species of tick-borne rickettsiae that have been validated so far belong to SFG. Furthermore, the members of SFG rickettsiae have been increasing as many new species have been proposed recently3,4,5,6,7.
In Japan, R. japonica was the first SFG Rickettsia discovered in 1984 as the causative agent of Japanese spotted fever (JSF)8,9. Since then, several other SFG rickettsiae, namely R. heilongjiangensis, R. helvetica, and R. tamurae have been recognised as etiological agents of human diseases10,11,12. SFG rickettsiae with unknown pathogenicity, such as R. asiatica and Candidatus R. tarasevichiae, have also been reported13,14. In addition, several studies conducted in Japan have documented the presence of other Rickettsia species/genotypes in animals and questing ticks15,16,17. However, in most cases, only single or a limited number of genes have been analysed, making it difficult to generate an overview of the genetic diversity of SFG rickettsiae, since multiple gene sequencing are recommended in the classification of rickettsial isolates18.
The relationship between SFG rickettsiae and their vector tick species has been studied previously. It is evident that some SFG rickettsiae, such as R. rickettsii, are associated with several different tick species in different genera, while others, such as R. conorii, are linked to specific tick species19. In Japan, R. japonica is considered to be in the former group since it has been recorded from wide range of tick species including Dermacentor taiwanensis, Haemaphysalis hystricis, H. cornigera, H. longicornis, H. flava, H. formosensis, H. megaspinosa, and Ixodes ovatus20. On the other hand, vector tick species of other rickettsiae, such as R. asiatica and R. heilongjiangensis, which are respectively transmitted by I. ovatus and H. concinna, seem to be limited11,13.
The aim of the present study was to understand the overall diversity of SFG rickettsiae and their vector tick species in Japan. By collecting questing ticks at more than 100 different sampling sites across Japan, a nationwide cross-sectional study for SFG rickettsiae was conducted. The samples included 19 different tick species covering most of the commonly found species in Japan. Our results indicate that there exist more SFG rickettsiae genotypes than previously known. The information on the relationship between SFG rickettsiae and vector ticks is useful for further characterisation of each rickettsiael member in more detail.
Results
Detection of SFG rickettsiae by real-time PCR for gltA
Out of 2,189 ticks, 373 (17.0%) samples were positive for Rickettsia spp. by citrate synthase gene (gltA) real-time PCR (Table 1). Among the 19 different tick species, seven tick species, namely D. taiwanensis, H. concinna, H. cornigera, H. yeni, I. pavlovskyi, I. tanuki, and I. turdus, were negative for rickettsiae infection. The highest infection rate was observed in I. nipponensis (80.0%), followed by H. longicornis (62.8%), I. monospinosus (58.6%), H. hystricis (57.8%), I. persulcatus (34.8%), A. testudinarium (23.5%), H. megaspinosa (17.4%), H. flava (10.2%), H. japonica (5.1%), H. kitaokai (4.1%), H. formosensis (2.8%), and I. ovatus (1.1%).
gltA genotyping
Out of 373 samples that tested positive for rickettsiae by real-time PCR for gltA, 352 samples yielded amplicons by conventional PCR for gltA, while 21 samples did not (Table 1). All the amplicons were successfully sequenced, which resulted in 15 different gltA genotypes (Fig. 1 and Table 1). In the present study, the gltA genotype is defined as a gltA sequence type that is different from the others even by a single nucleotide. The sequence alignment of all 15 gltA sequences is provided in Supplementary Fig. S1. All gltA genotypes (G1, G2, G6, G7, G9, G11, G12, G14, and G15) detected in the genus Haemaphysalis were clustered in the same clade, and five genotypes (G3, G4, G5, G10, and G13) obtained from the genus Ixodes were allocated to three different clusters while only one genotype (G8) was linked with the genus Amblyomma. (Fig. 1). A total of 13 genotypes were detected in only one single tick species, while two genotypes (G5 and G11) were detected in two different tick species: G5 was recovered from I. persulcatus and I. monospinosus, and G11 was from H. japonica and H. flava. Three tick species harboured multiple gltA genotypes: I. persulcatus, H. formosensis, and H. flava had 3, 3, and 2 different gltA genotypes, respectively.

A phylogenetic tree of spotted fever group rickettsiae based on the gltA gene sequences (537 bp). The analysis was performed using a maximum likelihood method with the Kimura 2-parameter model. All bootstrap values from 1,000 replications are shown on the interior branch nodes. The sequences detected in this study are indicated in red. The number of samples positive for each genotype is indicated in the parentheses. The simplified tick phylogeny consisting of 12 tick species is indicated on the top right. The colour highlights in the column indicate the presence of infections in each tick species.
Geographic information on gltA genotypes and host ticks
Table 2 represents the relationship between gltA genotypes and their geographical origins. Out of 15 genotypes, 11 genotypes (G1, G2, G3, G4, G5, G6, G8, G10, G11, G12, and G15) were detected from multiple geographical regions. The other 4 genotypes (G7, G9, G13, and G14) were detected in only one single tick species from a single region. G7 and G14 were detected in H. formosensis from Kyusyu region, while G9 and G13 were in H. kitaokai from Kansai region and I. ovatus from Tohoku region, respectively (Fig. 1). The present study employed H. formosensis and H. kitaokai from three different regions and I. ovatus collected from five different regions (see Supplementary Table S1).
Multiple genes sequencing
To further characterise Rickettsia spp. based on five other genes; outer membrane protein A gene (ompA), outer membrane protein B gene (ompB), 17-kDa common antigen gene (htrA), surface cell antigen-4 (sca4), and 16S ribosomal RNA gene (16S rRNA), PCR analyses were conducted on the selected samples of each gltA genotype. A total of 57 samples were employed in this analysis. We selected more than two samples from each genotype except for G7 which was found in only one sample (Table 3). The samples with higher rickettsial burden were selected based on the results of gltA real-time PCR. The mean rickettsial burden in the template DNA ranged from 2.3E + 2 to 2.1E + 4 copies/μL (Table 3). The htrA gene was successfully amplified and sequenced for all gltA genotypes. Although 16S rRNA PCR gave amplicons in all gltA genotypes, the following sequencing analysis revealed that rickettsial 16S rRNA gene sequences were obtained in only 12 gltA genotypes. The ompB, ompA and sca4 genes were amplified and sequenced in 11, five and six different gltA genotypes, respectively. All genes were successfully sequenced in two gltA genotypes (G6 and G7). Four genes were successfully amplified in six gltA genotypes (G1, G2, G5, G8, G10, and G11), and three genes were amplified in four gltA genotypes (G3, G4, G9, and G13). Only the htrA gene was amplified in three gltA genotypes (G12, G14, and G15) (Table 3). The sequencing analysis of the amplified products revealed that there were no sequence differences in any of the genes in the samples with the same gltA genotypes. The sequence types obtained from each gltA genotype were different from each other. The sequence identity with the closest Rickettsia species are provided in Supplementary Table S2.
Species classification of SFG rickettsiae
Phylogenetic trees inferred from ompA, ompB, sca4, htrA, and 16S rRNA analysis are shown in Fig. 2. G4 and G5 formed a distinct cluster with R. helvetica in all trees when sequences were available and thus were identified as R. helvetica. Being supported by more than three trees, G13, G10, G8, and G3 were identified as R. asiatica, R. monacensis (former Rickettsia sp. In56), and R. tamurae, and Candidatus R. tarasevichiae, respectively (Fig. 2). The other nine gltA genotypes could not be classified into specific species due to a lack of consensus between the trees and/or absence of sequences from previously validated rickettsial species in the same phylogenetic clusters.

Phylogenetic trees based on the sequences of the ompA (493 bp) (A), ompB (780 bp) (B), htrA (465 bp) (C), sca4 (887 bp) (D), and 16S rRNA (1,245 bp) (E) genes. The analyses were performed using a maximum likelihood method with the Kimura 2-parameter model. All bootstrap values from 1,000 replications are shown on the interior branch nodes. The sequences obtained in this study are shown in red.
Discussion
The present study included a total of 2,189 individual ticks collected at 114 different sampling sites in six regions of Japan for the screening of SFG rickettsiae. Our nationwide sampling enabled us to collect as many as 19 different tick species from four genera, most of which were common tick species prevalent in Japan. A first screening test using gltA real-time PCR revealed that 17.0% (373 out of 2,189) of the ticks were infected with SFG rickettsiae. This infection rate was comparative to the results of an earlier study where 21.9% (181 out of 827) of the ticks, including 10 different species collected from central (Shizuoka, Mie, and Wakayama prefectures) and southern (Kagoshima, Nagasaki, and Okinawa prefectures) parts of Japan, were positive for SFG rickettsiae16. Another nationwide survey conducted in 5 prefectures (Chiba, Hokkaido, Kochi, Tokushima, and Toyama prefectures) including JSF endemic areas reported an overall positive rate for SFG rickettsiae to be 25.8% (186 out of 722) in 10 different tick species21.
We determined partial sequences of the gltA gene of SFG rickettsiae by conventional PCR, which was previously designed to characterise SFG rickettsiae in Japan16. Based on the sequences of the gltA gene obtained from 352 ticks, the SFG rickettsiae detected in the present study were provisionally divided into 15 genotypes. In the molecular classification of SFG rickettsiae, the analysis of multiple genes commonly used by other researchers is a prerequisite18. Therefore, we attempted to obtain the sequences of five additional genes, ompA, ompB, htrA, sca4, and 16S rRNA. These efforts lead to the identification of four validated rickettsial species, namely R. asiatica, R. helvetica, R. monacensis, and R. tamurae, and the provisional species Candidatus R. tarasevichiae (Fig. 2).
Prior to this study, there was no official report of the presence of R. monacensis in Japan. A recent study indicated that Rickettsia sp. In56, a rickettsial strain reported from ticks in Japan21, might be a synonymous of R. monacensis22. Although several isolates of Rickettsia sp. In56 have been obtained from Japanese ticks23, lack of their sequence information prevents a direct comparison between Rickettsia sp. In56 and R. monacensis reported elsewhere. Nevertheless, the sequence analysis of multiple genes (gltA, ompA, ompB, htrA, and 16S rRNA) conducted in the present study confirmed the presence of R. monacensis in Japan (Figs 1 and 2). R. monacensis was initially isolated from I. ricinus collected from the English Garden in Germany using ISE6 cells24 and has been detected from the same tick species in Europe and neighbouring countries25,26,27,28,29. I. nipponensis and I. sinensis are considered as main vectors of R. monacensis in China and Korea, respectively30,31. In our study, R. monacensis was detected from four I. nipponensis samples collected in the Tohoku and Kansai regions, while none of the other tick species carried R. monacensis (Fig. 1 and Table 2). These results may suggest the relatively wide distribution of R. monacensis and a strong association of R. monacensis with I. nipponensis in Japan. This SFG rickettsiae caused Mediterranean spotted fever-like symptoms in humans in several countries32,33. More recently, the agent was isolated from the blood of a patient with an acute febrile illness in Korea22. Thus, clinicians should be aware of R. monacensis as a possible cause of non-JSF rickettsiosis in Japan.
Although we tried to characterise SFG rickettsiae with each prospective gltA genotype in further detail by sequencing five additional rickettsial genes, ompA, ompB, htrA, sca4, and 16S rRNA, the amplification was not successful for some genes (Table 3). The ompA and sca4 genes were amplified only from one third of the tested gltA genotypes. Considering the relatively high rickettsial abundance in the tested samples (Table 3), PCR failure is either because some of the SFG rickettsiae lack these genes as shown in TG rickettsiae that do not possess ompA gene34, or because there are nucleotide mismatches in the primer annealing sites. PCR failures of variable genes such as ompA, ompB, and sca4 are common issues in the genetic characterisation of SFG rickettsiae34,35. Thus further attempts including the development of universal primers and/or bacterial isolation followed by whole genome sequencing are required to determine the phylogenetic positions of uncharacterised Rickettsia spp.
In a previous nationwide survey of SFG rickettsiae conducted in Japan, Gaowa et al.16 classified the detected rickettsiae (n = 181) into five groups (groups 1–5) based on the gltA sequences16. Groups 1 and 2 were respectively identified as R. japonica and R. tamurae, whereas groups 3, 4, and 5, showing high sequence similarity with Rickettsia sp. LON-13, R. raoultii, and Candidatus R. principis, respectively, were not classified as validated rickettsial species16. In agreement with their report, we detected gltA sequences corresponding to groups 3 (G6), 4 (G2), and 5 (G1, G11, G12, G14, and G15) (Fig. 2). Unfortunately, limited information is available about these uncharacterised Rickettsia spp. In our study, G6 and G2 were respectively detected in H. longicornis and H. hystricis with high infection rates (62.8% and 57.8%, respectively) (Table 1), warranting further studies on the effect of these infections for the survival and reproductive fitness of their hosts.
We detected two gltA genotypes (G7 and G9), which were allocated into distinct clusters from Rickettsia spp. previously reported from Japan (Fig. 2). G7 and G9 showed the highest gltA sequence similarity with Rickettsia spp. reported from Kenya (KT257873) and Hungary (EU853834), respectively. Rickettsia sp. reported from Kenya was detected in Rhipicephalus maculatus36, while the one from Hungary was detected in H. inermis and provisionally named as Candidatus R. hungarica37. Since the sequences of other genes were not available from those Rickettsia spp., it was difficult to evaluate the degree of genetic relatedness in more detail. Nonetheless, the presence of closely related species in two geographically remote areas may indicate the worldwide distribution of these poorly characterised SFG rickettsiae. Since the present study provided the sequences of multiple genes of those rickettsiae, the information is useful in the classification of SFG rickettsiae.
In the present study, we found a strong association between rickettsial genotypes and their host tick species; 13 out of 15 gltA genotypes were detected in only one single tick species (Fig. 1 and Table 3). Furthermore, there was minimal geographical restriction for the 11 gltA genotypes that were recovered from multiple geographical regions (Table 2). These observations may indicate that most of the SFG rickettsiae species are found in ticks but not in vertebrate hosts in the natural environment. However, further examinations are needed to confirm this hypothesis by observing transstadial and transovarial transmission of these SFG rickettsiae in ticks. The effect of these rickettsial infections on tick physiology and reproduction remains to be elucidated.
Although the sampling was conducted at several JSF-endemic areas in Mie, Kagoshima, and Kumamoto prefectures, none of the ticks were infected with R. japonica. Considering the low level of genomic plasticity within R. japonica isolates38, it was hardly expected that a real-time PCR assay for gltA would result in false-negatives. The positive rate of R. japonica infection in the questing ticks was as low as 0.86% (18 out of 2,099), even in endemic areas as is the case in Shimane prefecture39. Collectively, the failure in detection of R. japonica might be partly attributed to the sample selection procedure with which only a maximum of 10 individual ticks per species, stage/sex, and site were tested for SFG rickettsiae infection. Therefore, it should be noted that the present study might not fully disclose the diversity of SFG rickettsiae in Japan, which warrants further investigations by employing a larger number of samples.
The present study has extended our knowledge on the diversity of SFG rickettsiae prevalent in Japan. In addition to previously recognised rickettsial species such as R. asiatica, R. helvetica, R. monacensis (formerly reported as Rickettsia sp. In56), R. tamurae, and Candidatus R. tarasevichiae, several uncharacterised Rickettsia spp. including ones showing high similarities with those designated as novel Rickettsia spp. detected in geographically remote countries such as Kenya and Hungary were discovered. A strong association between rickettsial genotypes and their host ticks suggests a long-term relationship between SFG rickettsiae and ticks. Further investigations on the potential roles of these SFG rickettsiae on ticks are required to understand the mechanisms underlying widespread existence of genetically variable rickettsiae in ticks.
Materials and Methods
Sample collection
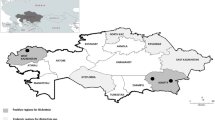
Ticks were collected by flagging a flannel cloth over the vegetation during the period of tick activity (between April 2013 and March 2016) at 114 different sampling sites in 12 different prefectures. The sampling sites were categorised into geographical blocks: Hokkaido (Hokkaido prefecture), Tohoku (Yamagata and Fukushima prefectures), Chubu (Nagano and Shizuoka prefectures), Kansai (Mie, Nara, and Wakayama prefectures), Kyushu (Kumamoto, Miyazaki, and Kagoshima prefectures), and Okinawa (Okinawa prefecture) (Fig. 3). All field-collected ticks were transferred to small Petri dishes and preserved in an incubator at 16 °C until use.

Sample collection sites.
Tick species identification
Tick species were identified morphologically using standard keys under a stereomicroscope40,41. When more than 10 ticks with the same species and stage/sex were collected from the same sampling sites, a maximum of 10 individual ticks were analysed per species, stage/sex and site. A total of 2,189 individuals (103 nymphs and 2,086 adults) in four genera were examined in this study. These included one species in the genus Amblyomma (A. testudinarium, n = 85), one species in the genus Dermacentor (D. taiwanensis, n = 12), 10 species in the genus Haemaphysalis (H. concinna, n = 7; H. cornigera, n = 1; H. flava, n = 128; H. formosensis, n = 253; H. japonica, n = 78; H. hystricis, n = 64; H. kitaokai, n = 74; H. longicornis, n = 86; and H. megaspinosa, n = 201; H. yeni, n = 1) and 7 species in the genus Ixodes (I. monospinosus, n = 58; I. nipponensis, n = 5; I. ovatus, n = 652; I. pavlovskyi, n = 33; I. persulcatus, n = 446; I. tanuki, n = 2; and I. turdus, n = 3). Out of 2,189 ticks, 975, 1,111, and 103 were male, female, and nymph, respectively.
DNA extraction
Ticks were individually washed with 70% ethanol followed by washing with sterile PBS twice, then homogenised in 100 μL of high glucose Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Life Technologies) by using Micro Smash MS-100R (TOMY, Tokyo, Japan) for 30 sec at 3,000 rpm as described previously42. DNA was extracted from 50 μL of the tick homogenate using a blackPREP Tick DNA/RNA Kit (Analytikjena, Germany) according to the manufacturer’s instructions, while the other half was kept at −80 °C for future bacterial isolation.
Real-time PCR
All samples were first screened for rickettsial gltA using real-time PCR to detect SFG and TG rickettsiae as described previously43. The primers and probes used are shown in Table 4. Reactions were performed in a 20.0 μL of reaction mixture containing 10.0 μL of THUNDERBIRD Probe qPCR Mix (Toyobo, Osaka, Japan), 300 nM of each primer, 200 nM of probe, 5.0 μL of template DNA, and distilled water. The reaction was carried out in a C1000 Thermal Cycler with a CFX96 Real-Time PCR Detection System (BioRad Laboratories, Hercules, CA) at conditions of 50 °C for 3 min, 95 °C for 1 min, and 40 cycles of 95 °C for 15 sec and 60 °C for 1 min. Each run included a blank control and serially diluted plasmid standards (106, 104, and 102 copies/reaction) as described previously35.
Conventional PCR
All the samples that were positive for gltA by real-time PCR were further characterised by conventional PCR targeting an approximately 580 bp sequence of the gltA gene using the primers gltA_Fc and gltA_Rc (Table 4)16. The PCR was carried out in a 25.0 μL reaction mixture containing 12.5 μL of 2 × KAPA blood PCR Kit (KAPA Biosystems, USA), 200 nM of each primer, 2.0 μL of DNA template, and sterile water. The reactions were performed at 95 °C for 5 min; followed by 45 cycles of 95 °C for 30 sec, 55 °C for 30 sec, and 72 °C for 40 sec; and 72 °C for 5 min. PCR products were electrophoresed at 100 V in a 1.2% agarose gel for 25 min. DNA from the R. japonica YH strain and sterile water were included in each PCR run as positive and negative controls, respectively. For the selected samples from each gltA genotype (n = 57), additional PCR assays were conducted based on five genes: ompA, ompB, sca4, htrA, and 16S rRNA. The primer sets used for each assay are shown in Table 444,45,46,47,48. PCR conditions were the same as mentioned above except for the annealing temperature (48 °C for ompA and ompB PCRs, 52 °C for 16S rRNA and htrA PCRs, and 50 °C for sca4 PCR).
Sequencing
The amplified PCR products were purified using a Wizard® SV Gel and PCR Clean-Up System Kit (Promega, USA). Sanger sequencing was conducted using the BigDye Terminator version 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and an ABI Prism3130x genetic analyser according to the manufacturers’ instructions. The sequences data were assembled using ATGC software version 6.0.4 (GENETYX, Tokyo, Japan). The sequences obtained were submitted to the DNA Data Bank of Japan (DDBJ) (http://www.ddbj.nig.ac.jp) under accession numbers (gltA: LC379427-LC379443; ompA: LC379461-LC379465; ompB: LC379466-LC379476; htrA: LC379444-LC379460; sca4: LC379477-LC379482; 16S rRNA: LC379483-LC379494).
Phylogenetic analysis
The nucleotide sequences obtained were aligned with representative sequences of known rickettsial species available on GenBank using ClustalW 1.6 as implemented in MEGA 749. After manual modification of the alignments, phylogenetic trees were constructed using the maximum likelihood method using Kimura 2-parameter with bootstrap tests of 1,000 replicates via MEGA. R. bellii was included as an outgroup for the bases of the trees for gltA, ompB, and htrA, while R. typhi, R. akari, and Ehrlichia chaffeensis were used as outgroups for sca4, ompA, 16S rRNA, respectively. In order to generate a phylogenetic tree of tick species that was positive for Rickettsia spp., partial nucleotide sequences of mitochondrial 16S rRNA gene obtained from GenBank were used.
Data Availability
All data discussed in the manuscript is included in the paper.
References
Raoult, D. & Roux, R. Rickettsioses as paradigms of new or emerging infectious diseases. Clin. Mirobiol. Rev. 10, 694–719 (1997).
Gillespie, J. J. et al. Plasmids and rickettsial evolution: insight from Rickettsia felis. PLoS One. 2, e266, https://doi.org/10.1371/journal.pone.0000266 (2007).
Karpathy, S. E., Slater, K. S., Goldsmith, C. S., Nicholson, W. L. & Paddock, C. D. Rickettsia amblyommatis sp. nov., a spotted fever group Rickettsia associated with multiple species of Amblyomma ticks in North, Central and South America. Int. J. Syst. Evol. Microbiol. 66, 5236–5243 (2016).
Abdad, M. Y. et al. Rickettsia gravesii sp. nov.: a novel spotted fever group Rickettsia in Western Australian Amblyomma triguttatum ticks. Int. J. Syst. Evol. Microbiol. 67, 3156–3161 (2017).
Dall’Agnol, B. et al. Candidatus Rickettsia asemboensis in Rhipicephalus sanguineus ticks, Brazil. Acta Trop. 167, 18–20 (2017).
Lee, J. K. et al. Rickettsia parkeri and Candidatus Rickettsia andeanae in questing Amblyomma maculatum (Acari: Ixodidae) from Mississippi. J. Med. Entomol. 54, 476–480 (2017).
Moreira-Soto, R. D., Moreira-Soto, A., Corrales-Aguilar, E., Calderon-Arguedas, O. & Troyo, A. Candidatus Rickettsia nicoyana: A novel Rickettsia species isolated from Ornithodoros knoxjonesi in Costa Rica. Ticks Tick Borne Dis. 8, 532–536 (2017).
Uchida, T., Tashiro, F., Funato, T. & Kitamura, Y. Isolation of a spotted fever group Rickettsia from a patient with febrile exanthematous illness in Shikoku, Japan. Microbiol. Immunol. 30, 1323–1326 (1986).
Mahara, F. Japanese spotted fever: Report of 31 cases and review of the literature. Emerg. Infect. Dis. 3, 105–111 (1997).
Noji, Y. et al. The first reported case of spotted fever in Fukui Prefecture, the Northern part of Central Japan. Jpn. J. Infect. Dis. 58, 112–114 (2005).
Ando, S. et al. Human Rickettsia heilongjiangensis infection, Japan. Emerg. Infect. Dis. 16, 1306–1308 (2010).
Imaoka, K., Kaneko, S., Tabara, K., Kusatake, K. & Morita, E. The first human case of Rickettsia tamurae infection in Japan. Case Rep. Dermatol. 3, 68–73 (2011).
Fujita, H., Fournier, P. E., Takada, N., Saito, T. & Raoult, D. Rickettsia asiatica sp. nov., isolated in Japan. Int. J. Syst. Evol. Microbiol. 56, 2365–2368 (2006).
Inokuma, H., Ohashi, M., Jilintai, Tanabe, S. & Miyahara, K. Prevalence of tick-borne Rickettsia and Ehrlichia in Ixodes persulcatus and Ixodes ovatus in Tokachi District, Eastern Hokkaido, Japan. J. Vet. Med. Sci. 69, 661–664 (2007).
Baba, K., Kaneda, T., Nishimura, H. & Sato, H. Molecular detection of spotted fever group Rickettsia in feral raccoons (Procyon lotor) in the Western Part of Japan. J. Vet. Med. Sci. 75, 195–197 (2013).
Gaowa et al. Rickettsiae in ticks, Japan, 2007–2011. Emerg. Infect. Dis. 19, 338–340 (2013).
Someya, A., Ito, R., Maeda, A. & Ikenaga, M. Detection of rickettsial DNA in ticks and wild boars in Kyoto City, Japan. J. Vet. Med. Sci. 77, 37–43 (2015).
Fournier, P. E. et al. Gene sequence based criteria for identification of new Rickettsia isolates and description of Rickettsia heilongjiangensis sp. nov. J. Clin. Microbiol. 41, 5456–5465 (2003).
Socolovschi, C., Mediannikov, O., Raoult, D. & Parola, P. The relationship between spotted fever group rickettsiae and ixodid ticks. Vet. Res. 40, 34 (2009).
Ando, S. & Fujita, H. Diversity between spotted fever group Rickettsia and ticks as vector. Eisei Dobutsu. 64, 5–7, https://doi.org/10.7601/mez.64.5 (2013).
Ishikura, M. et al. Phylogenetic analysis of spotted fever group rickettsiae based on gltA, 17-kDa, and rOmpA genes amplified by nested PCR from ticks in Japan. Microbiol. Immunol. 47, 823–832 (2003).
Kim, Y. S. et al. First isolation of Rickettsia monacensis from a patient in South Korea. Microbiol. Immunol. 61, 258–263 (2017).
Fujita, H. et al. Tick species and tick-borne rickettsiae confirmed by the year of 2012 in Fukushima prefecture, Japan. Med. Entomol. Zool. 64, 37–41 (2013).
Simser, J. A. et al. Rickettsia monacensis sp. nov., a spotted fever group Rickettsia, from ticks (Ixodes ricinus) collected in a European City Park. Appl. Environ. Microbiol. 68, 4559–4566 (2002).
Sréter-Lancz, Z., Sréter, T., Széll, Z. & Egyed, L. Molecular evidence of Rickettsia helvetica and R. monacensis infection in Ixodes ricinus from Hungary. Ann. Trop. Med. Parasitol. 99, 325–330 (2005).
Milhano, N. et al. Coinfections of Rickettsia slovaca and Rickettsia helvetica with Borrelia lusitaniae in ticks collected in a Safari Park, Portugal. Ticks Tick Borne Dis. 1, 172–177 (2010).
Špitalská, E., Boldiš, V., Derdáková, M., Selyemová, D. & Taragel’ová, V. R. Rickettsial infection in Ixodes ricinus ticks in urban and natural habitats of Slovakia. Ticks Tick Borne Dis. 5, 161–165 (2014).
Venclikova, K., Rudolf, I., Mendel, J., Betasova, L. & Hubalek, Z. Rickettsiae in questing Ixodes ricinus ticks in the Czech Republic. Ticks Tick Borne Dis. 5, 135–138 (2014).
Biernat, B., Stańczak, J., Michalik, J., Sikora, B. & Cieniuch, S. Rickettsia helvetica and R. monacensis infections in immature Ixodes ricinus ticks derived from sylvatic passerine birds in west-central Poland. Parasitol. Res. 115, 3469–3477 (2016).
Ye, X. et al. Vector competence of the tick Ixodes sinensis (Ascari: Ixodidae) for Rickettsia monacensis. Parasit. Vectors. 7, 512, https://doi.org/10.1186/s13071-014-0512-8 (2014).
Shin, S. H. et al. Detection of Rickettsia monacensis from Ixodes nipponensis collected from rodents in Gyeonggi and Gangwon Provinces, Republic of Korea. Exp. Appl. Acarol. 61, 337–347 (2013).
Jado, I. et al. Rickettsia monacensis and human disease, Spain. Emerg. Infect. Dis. 13, 1405–1407 (2007).
Madeddu, G. et al. Rickettsia monacensis as cause of Mediterranean spotted fever-like illness, Italy. Emerg. Infect. Dis. 18, 702–704 (2012).
Ngwamidiba, M., Blanc, G., Raoult, D. & Fournier, P. E. Sca1, a previously undescribed paralog from autotransporter protein-encoding genes in Rickettsia species. BMC Microbiol. 6, 12, https://doi.org/10.1186/1471-2180-6-12 (2006).
Nakao, R. et al. High prevalence of spotted fever group rickettsiae in Amblyomma variegatum from Uganda and their identification using sizes of intergenic spacers. Ticks Tick Borne Dis. 4, 506–512 (2013).
Mwamuye, M. M. et al. Novel Rickettsia and emergent tick-borne pathogens: A molecular survey of ticks and tick-borne pathogens in Shimba Hills National Reserve, Kenya. Ticks Tick Borne Dis. 8, 208–218 (2017).
Hornok, S. et al. Molecular investigation of hard ticks (Acari: Ixodidae) and fleas (Siphonaptera: Pulicidae) as potential vectors of rickettsial and mycoplasmal agents. Vet. Microbiol. 140, 98–104 (2010).
Akter, A. et al. Extremely low genomic diversity of Rickettsia japonica distributed in Japan. Genome Biol. Evol. 9, 124–133 (2017).
Tabara, K. et al. High incidence of rickettsiosis correlated to prevalence of Rickettsia japonica among Haemaphysalis longicornis tick. J. Vet. Med. Sci. 73, 507–510 (2011).
Yamaguti N, Tipton V. J., Keegan H. L. & Toshioka S. Ticks of Japan, Korea and the Ryushu islands. Brigham Young University Science Bulletin, Biological Series 15 1971.
Nakao, M., Miyamoto, K. & Kitaoka, S. A new record of Ixodes pavlovskgi Pomerantzev from Hokkaido, Japan (Acari: Ixodidae). Eisei Dobutsu. 43, 229–234, https://doi.org/10.7601/mez.43.229 (1992).
Nakao, R., Magona, J. W., Zhou, L., Jongejan, F. & Sugimoto, C. Multi-locus sequence typing of Ehrlichia ruminantium strains from geographically diverse origins and collected in Amblyomma variegatum from Uganda. Parasit. Vectors. 4, 137, https://doi.org/10.1186/1756-3305-4-137 (2011).
Stenos, J., Graves, S. R. & Unsworth, N. B. A highly sensitive and specific real-time PCR assay for the detection of spotted fever and typhus group rickettsiae. Am. J. Trop. Med. Hyg. 73, 1083–1085 (2005).
Regnery, R. L., Spruill, C. L. & Plikaytis, B. D. Genotypic identification of rickettsiae and estimation of intraspecies sequence divergence for portions of two rickettsial genes. J. Bacteriol. 173, 1576–1589 (1991).
Roux, V. & Raoult, D. Phylogenetic analysis of members of the genus Rickettsia using the gene encoding the outer-membrane protein rOmpB (ompB). Int. J. Syst. Evol. Microbiol. 50, 1449–1455 (2000).
Sekeyova, Z., Roux, V. & Raoult, D. Phylogeny of Rickettsia spp. inferred by comparing sequences of gene D, which encodes an intracytoplasmic protein. Int. J. Syst. Evol. Microbiol. 51, 1353–1360 (2001).
Labruna, M. B. et al. . Rickettsia belli and Rickettsia amblyommii in Amblyomma ticks from the State of Rondonia, Western Amazon, Brazil. J. Med. Entomol. 41, 1073–1081 (2004).
Anstead, C. A. & Chilton, N. B. A novel Rickettsia species detected in vole ticks (Ixodes angustus) from Western Canada. Appl. Environ. Microbiol. 79, 7583–7589 (2013).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Acknowledgements
We would like to thank to all collaborators who supported in collection of ticks in each prefecture, especially Mrs. Ryuji Yamamoto and Kazuya Okuno in Wakayama prefecture. We are also grateful to Dr. Tsuneo Uchiyama (The University of Tokushima Graduate School, Tokushima, Japan) for providing R. japonica YH strain DNA. This work was financially supported by JSPS KAKENHI Grant-in-Aid for Young Scientists (B) (16K19112) and (A) (15H05633) and for Scientific Research on Innovative Areas (16H06429, 16K21723, and 16H06431). Funding was also provided in part by the Agency for Medical Research and Development (AMED) and Japan International Cooperation Agency (JICA) within the framework of the Science and Technology Research Partnership for Sustainable Development (SATREPS) (17jm0110005h0006).
Author information
Authors and Affiliations
Contributions
R.N. and M.J.T. designed the study and wrote the manuscript. M.J.T., Y.Q., M.K. and A.M.K. performed the experiments. K.M., M.K., A.M.K., N.M., K.C., J.S., M.G., M.A. and H.O. collected the samples. R.O., K.K., A.T., C.S. and N.I. contributed the study design. All authors wrote and agreed the final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thu, M.J., Qiu, Y., Matsuno, K. et al. Diversity of spotted fever group rickettsiae and their association with host ticks in Japan. Sci Rep 9, 1500 (2019). https://doi.org/10.1038/s41598-018-37836-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37836-5
This article is cited by
-
Summer collection of multiple southern species of ticks in a remote northern island in Japan and literature review of the distribution and avian hosts of ticks
Experimental and Applied Acarology (2023)
-
A case report of Rickettsia-like infection in a human patient from Slovakia
Biologia (2022)
-
Genetic diversity of Rickettsia africae isolates from Amblyomma hebraeum and blood from cattle in the Eastern Cape province of South Africa
Experimental and Applied Acarology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
