
Abstract
Ketogulonicigenium vulgare has been widely used in vitamin C two-step fermentation. Four K. vulgare strains (WSH-001, Y25, Hbe602 and SKV) have been completely genome-sequenced, however, less attention was paid to elucidate the reason for the differences in 2-KGA yield on genetic level. Here, a novel K. vulgare SPU B805 with higher 2-keto-L-gulonic acid (2-KGA) yield, was genome-sequenced to confirm harboring one circular chromosome with plasmid free. Comparative genome analyses showed that the absence of plasmid 2 was an important factor for its high 2-KGA productivity. The amino acid biosynthetic pathways in strain SPU B805 are much more complete than those in other K. vulgare strains. Meanwhile, strain SPU B805 harbored a complete PPP and TCA route, as well as a disabled EMP and ED pathway, same as to strain SKV, whereas strain WSH-001, Y25 and Hbe602 harbored complete PPP, ED, TCA pathway and a nonfunctional EMP pathway. The transcriptome of strain SPU B805 validated the carbon metabolism in cytoplasm mainly through the PPP pathway due to its higher transcriptional levels. This is the first time to elucidate the underlying mechanism for the difference in 2-KGA yield, and it is of great significance for strain improvement in the industrial fermentation.
Similar content being viewed by others
Introduction
Ketogulonicigenium vulgare has been widely used for the bioconversion of L-sorbose to 2-keto-L-gulonic acid (2-KGA), the key precursor of vitamin C in the industrial production. In this process, K. vulgare is usually co-cultured with companion strain, Bacillus megatherium, which is generally considered to provide nutrients or metabolic substances for the growth of K. vulgare1. The known 2-KGA-producing strains, K. vulgare Y25, K. vulgare WSH-001, K. vulgare SKV and K. vulgare Hbe602 have been completely genome-sequenced and reported2,3,4,5. The genome-scale metabolic model of K. vulgare WSH-001 was reconstructed and analyzed by flux balance analysis (FBA)6. Total 116 genes, located on the chromosome, were identified as the essential genes for the growth of K. vulgare WSH-0016. Among them, approximately half of the essential genes participated in the metabolism of nucleotides and amino acids. Some specific amino acids significantly affected the growth of cells and the accumulation of 2-KGA7,8; moreover, the uptake rates of amino acids had a linear positive correlation with cell growth rate6. Additional, K. vulgare WSH-001 had a powerful transporting system to absorb nutrients and various amino acids3. Although many researches on metabolic pathways have been carried out, the understanding of the K. vulgare is still a partial picture due to its unique growth characters. Therefore, it is necessary to discover the panorama of whole metabolism features of different 2-KGA-producing strains.
In prokaryotes, sugar has a variety of oxidation decomposition pathways, the most common of which are the Embden-Meyerhoff-Parnass (EMP) pathway, the Entner-Doudoroff (ED) pathway and the pentose phosphate pathway (PPP)9. However, these routes are different in the ATP yield, cofactors and chemical intermediates. The EMP pathway is a process by which glucose is not completely oxidized to pyruvate under anaerobic conditions10. In the classical EMP pathway, glucose is phosphorylated twice and cleaved into glyceraldehyde 3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP), and then two G3P are formed by the reversible interconversion between DHAP and G3P. G3P is metabolized to pyruvate in the subsequent glycolysis and coupled with ATP production through the substrate-level phosphorylation. So, one mole of glucose is oxidized to produce a net of two moles of ATP and two moles of NADH through the EMP pathway.
The ED pathway is a wildly distributed glucose catabolic route in nature11,12. In the ED pathway, glucose is phosphorylated to form glucose 6-phosphate (G6P), and then oxidized to 6-phosphogluconate (6PG) by glucose-6-phosphate dehydrogenase (Zwf)11. 6PG is then oxidized to 2-keto-3-deoxy-6-phosphogluconic acid (KDPG) by KDPG aldolase (Eda), which is then cleaved to one pyruvate and one G3P. G3P is used to produce ATP through substrate-level phosphorylation in latter glycolysis. KDPG is exclusively generated in the ED pathway and Eda is the key enzyme unique to the ED pathway10. The ED pathway can achieve the same conversion rate as the EMP pathway do, but fewer enzymes are required10. During the process, only one mole of ATP as well as one mole of NADH and one mole of NADPH are produced when one mole of glucose is decomposed.
The PPP pathway, as a fundamental component in the carbon metabolism, can be divided into the oxidative branch and non-oxidative branch13. In the oxidative branch, G6P is metabolized into ribulose 5-phosphate (Ru5P), NADPH and carbon dioxide by the enzyme Zwf and 6-phosphate gluconate dehydrogenase (Gnd). In the non-oxidative branch, the glycolytic intermediates fructose 6-phosphate (F6P), G3P and sedoheptulose 7-phosphate (S7P) are rearranged to generate ribose 5-phosphate (R5P) and other sugar phosphate for the subsequent biosynthesis of nucleic acids and amino acids13. Zwf plays an important role in the fine-tuning of PPP pathway, but it is not the unique to the PPP pathway because of its product 6PG can be channeled into ED pathway for further metabolism11. Unlike the EMP and ED pathway, one mole of glucose is decomposed to produce a net of two moles of NADPH through the PPP pathway.
Although several strains of K. vulgare have been genome-sequenced, annotated and published2,3,4,5, but their attentions were mainly focused on the elucidation of the metabolic pathway of 2-KGA biosynthesis and symbiosis mechanism with companion strain4,14. Less attention was paid to explore the mechanism of different 2-KGA yield on the genetic level in different strains. Therefore, in this work we studied the differences in genome and carbon metabolism pathway between K. vulgare SPU B805 and the four reported K. vulgare strains to understand the variations in the growth and 2-KGA production, as well as the advantages of K. vulgare SPU B805. K. vulgare SPU B805, a new 2-KGA-producing strain, contains only a circular chromosome without any plasmid. The COG and RAST annotation revealed that the genes encoding gluconate 2-dehydrogenase (GA2DH), one of L-sorbosone dehydrogenase (SNDH) and the key enzymes (Eda and Edd) unique to the ED pathway were all absent from the genome of strain SPU B805, which may be the underling mechanism that contributes to its high 2-KGA productivity. The complete pathways of glycine, serine, threonine and proline in SPU B805, whereas absent from other K. vulgare strains, may be another reason for high 2-KGA yield of SPU B805. Meanwhile, the comparative genome analysis showed that strain SPU B805 and SKV contained a complete PPP and TCA cycle, as well as a disabled EMP and ED pathway. Whereas, strain WSH-001, Y25 and Hbe602 harbor complete PPP, ED, TCA pathways and a nonfunctional EMP pathway. The transcriptome of strain SPU B805 validated that the PPP pathway was of major importance for cytoplasmic carbon oxidation. Our findings provide a complementary platform for understanding the underlying mechanism for the different central carbon metabolic features and 2-KGA yield of different K. vulgare strains. It also provides a useful framework for further physiology and gene engineering research. This is of great significance for K. vulgare strain improvement and genetic engineering in the 2-KGA industrial fermentation.
Results
2-KGA yield of K. vulgare SPU B805 in the co-culture system
K. vulgare SPU B805 was fermented with B. megatherium SPU B806 in 5-L bioreactor for 2-KGA production. The 2-KGA production was 82.74 ± 0.77 g/L with a 95.97% conversion rate at the end of the fermentation, and the absorbance at 660 nm was 5.63 ± 0.17, which means the cell concentration is 52 ± 1.4 × 108 CFU/mL (Table 1; Supplementary Fig. S1). Because no fermentation data of Y25 and SKV, the 2-KGA production abilities were compared between Hbe602, WSH-001 and SPU B805. As shown in Table 1, strain SPU B805 showed significantly differences (p < 0.001) compared with K. vulgare Hbe602 in the 2-KGA yield (increased by 25.55%, increment of 16.84 mg/ml), productivity (increased by 241.82%), 2-KGA conversion rate (increased by 25.55%) and fermentation cycle (decreased by 172.73%); similarly, strain SPU B805 showed significantly differences (p < 0.001) compared with K. vulgare WSH-001 in the 2-KGA yield (increased by 23.81%, increment of 15.91 mg/ml.), productivity (increased by 102.15%), 2-KGA conversion rate (increased by 8.48%) and fermentation cycle (decreased by 59.09%). The cell growth (OD660) of SPU B805 was also significantly higher than that of WSH-001 (p < 0.05). These superior qualities aroused our great interest to understand the mechanism. So, the genome of K. vulgare SPU B805 was sequenced, annotated and compared with all of the reported 2-KGA-producing strains (SKV, Hbe602, WSH-001 and Y25).
Genomic features of K. vulgare SPU B805
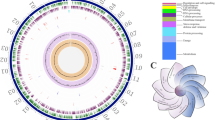
The complete genome of K. vulgare SPU B805 was sequenced and deposited in the GenBank database with the accession number CP017622. The genome schema was drawn as Supplementary Fig. S2. It is 3,032,608 bp in length with a G + C content of 61.7%. Based on 16S rRNA gene phylogenetic analysis, K. vulgare SPU B805 is highly homologous to a reported 2-KGA-producing strain of K. vulgare SKV5 (see Supplementary Fig. S3). The comparison of K. vulgare SPU B805 and K. vulgare Y252, K. vulgare WSH-0013, K. vulgare SKV5 and K. vulgare Hbe6024 in genome characteristics was summarized in Table 2. K. vulgare SPU B805 has the largest genome among all the strains, but it doesn’t encode the most genes and proteins. No plasmid is found in K. vulgare SPU B805, while other four strains have 1–2 plasmids. The number of rRNA (15) and tRNA (58) are the same as those in other strains except for tRNA (59) in WSH-001. The number of sorbose dehydrogenase (sdh) and sorbosone dehydrogenase (sndh), key genes responsible for the bioconversion of L-sorbose to 2-KGA, is different among all the strains, shown in Table 2. There are 5 sdh genes (KVC_2764, KVC_2744, KVC_0718, KVC_1927, KVC_0337) and 1 sndh gene (KVC_0605) in strain SPU B805.
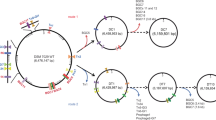
Furthermore, the genome-scale sequence comparison conducted by LAST software showed that the genome of K. vulgare SPU B805 is more similar with K. vulgare SKV (see Supplementary Fig. S4). Strain SPU B805, showing a 99% identity to strain SKV, harbors one circular chromosome without any plasmid. However, strain SKV contains one circle chromosome and one circle plasmid, while the other strains (Y25, WSH-001, Hbe602) consist of one circle chromosome and two circle plasmids, respectively. Therefore, it is the first time to find a 2-KGA-producing strain without any plasmid. The frame diagram of genome comparison between K. vulgare SPU B805, SKV, Hbe602, WSH-001 and Y25 was shown in Fig. 1. Most of the genome sequences are highly consistent except for some insertion, dislocation and rearrangement. Further analysis revealed that the DNA fragment of plasmid 1 (for example, namely as plasmid pKvSKV1 in SKV) was completely inserted into the circular chromosome (from 125,709 bp to 393,697 bp) of SPU B805, while the DNA fragment of plasmid 2 (belonging to strain Hbe602, WSH-001 and Y25), was completely lost in SPU B805.

The genome comparison of K. vulgare SPU B805 with the published genomes. The frames of the same color represent the homologous genome fragments. The frames under the main body represent the inverted fragments. The sequence of plasmid pKvSKV1 (belong to SKV), plasmid 1 (belong to Hbe602), pKVU_100 (belong to WSH-001) and pYP1 (belong to Y25) with highly similarity is inserted into the genome of K. vulgare SPU B805 (dark grey parts), while plasmid 2 (belong to SKV) or pKVU_200 (belong to WSH-001) or pYP12 (belong to Y25) (white fragments) is missing in the K. vulgare SPU B805 genome.
Genome annotation of K. vulgare SPU B805
To facilitate the gene function analysis, the genes on chromosome and plasmid 1 of the four published K. vulgare strains (SKV, Hbe602, WSH-001 and Y25) were selected to compare the distribution of COG classification with SPU B805 (see Supplementary Fig. S5). Consequently, in SPU B805, the number of genes related to amino acid transport and metabolism (E) is similar to that of Hbe602 and Y25 and higher than that in SKV and WSH-001. These functional genes encode many transporters, which absorb nutrients from the environment to compensate for its metabolic defects. Besides, the number of genes related to inorganic ion transport and metabolism (P) and translation, ribosomal structure and biogenesis (J) in SPU B805 is more than those of the reported K. vulgare strains. Inorganic ions are a kind of important cofactor involved in various physiological reactions in cell. The strong translation ability may be helpful to biosynthesize protein in K. vulgare SPU B805.
On the basis of the COG and RAST annotation, the genes on plasmid 2 of strain Hbe602, WSH-001 and Y25 account for about 7% of the genome. A majority of them are related to amino acid transport and metabolism (E), and assigned as “an ABC transporter” (Supplementary Fig. S5 and Table S1). Besides, some of the genes for carbohydrate metabolism are annotated as gluconate and ketogluconate metabolism. For example, the enzyme gluconate 2-dehydrogenase (GA2DH) can convert 2-KGA into idonate, followed flowing to 6-phosphogluconate (6PG) and subsequently entering the PPP for energy and biomass production6, finally decrease the carbon flow to 2-KGA and lower the 2-KGA production. Additionally, one of the L-sorbosone dehydrogenase (SNDH) genes is also found to be located on plasmid 2. The over-expression of SNDH (encoded by plasmid 2) in K. vulgare Hbe602 produced an obvious byproduct (not identified its chemical structure in the published paper), so as to decrease the 2-KGA yield15. Moreover, the key genes unique to ED pathway, 2-keto-3-deoxy-6-phosphogluconate aldolase (Eda) and phosphogluconate dehydratase (Edd), are annotated to be located on plasmid 2. The deletion of edd and eda genes from Gluconobacter oxydans 621H resulted in a lower overall sugar uptake by cells in the cytoplasm, and led to more sugar being left in the periplasm and converted to the end-product (2-keto-gluconate) with high yield16. The loss of ED pathway maybe has a similar effect on 2-KGA production in K. vulgare. Fortunately, these genes (GA2DH, SNDH, Eda and Edd) located on the indigenous plasmid 2, are all absent from the genome of K. vulgare SPU B805, which may be a reason for its high 2-KGA production. Therefore, presumably, elimination of the above genes or indigenous plasmid 2 is an effective method to enhance 2-KGA production by K. vulgare. Moreover, to construction an XFP-PTA pathway as described by Wang, et al.17 to decrease the carbon flux towards ED pathway with lower carbon loss is another pathway to promote the 2-KGA yield.
Moreover, a total of 2933 coding sequences were obtained by RAST annotation in strain SPU B805, which belonged to 26 subsystems, including cofactor, membrane transport metabolism, nucleotides metabolism, protein metabolism, regulation and cell signaling, amino acids and carbohydrate metabolism, and so on. Among them, genes regarding to amino acid metabolism were accounted for the highest proportion (15%) (Fig. 2 and Supplementary Table S2). However, the genes coding histidinol-phosphate (EC 3.1.3.15), leucine-alanine transaminase (EC 2.6.1.12), alanine transaminase (EC 2.6.1.2) and asparagine synthetase (EC 6.3.1.1) were found to be absent in strain SPU B805, which resulted in the incomplete biosynthesis pathway of histidine, alanine and asparagine (Fig. 3). While in strain WSH-001, the biosynthesis pathways of more amino acids, such as histidine, glycine, lysine, proline, threonine, methionine, leucine and isoleucine, were deficient due to absence of one or more key enzymes7. So the biosynthetic pathways of amino acid in K. vulgare SPU B805 are more complete than those in the reported K. vulgare strain WSH-001. Previous study showed that glycine, serine, threonine and proline were important factors for 2-KGA biosynthesis, and threonine affected cell growth significantly8. Glycine and serine are donors of one-carbon units in one-carbon metabolism, and threonine can be converted into glycine and acetyl-CoA. The impairment of these amino acids biosynthesis pathways will weaken the biosynthesis of nucleic acid and protein, and the biogenesis methyl group. Moreover, proline can decrease the destruction of microbial cells resulted by the osmotic stress at high 2-KGA concentrations18. Therefore, presumably, the more complete biosynthesis pathways in strain SPU B805 may contribute to the growth of K. vulgare and the accumulation of 2-KGA. Besides, the membrane transport genes were abundant in strain SPU B805, which might help to transport and absorb the nutrients and metabolites released by B. megatherium1.

Gene distributions of metabolic subsystems of K. vulgare SPU B805.

Reconstruction of the amino acid biosynthesis pathway of K. vulgare. Red ovals and dark red ovals represent the complete and incomplete amino acids biosynthesis pathway of K. vulgare SPU B805. The blue ovals represent the incomplete biosynthesis pathway of amino acids of K. vulgare WSH-001. The dotted lines indicate the genes related to the amino acid biosynthesis are absent.
Construction of carbohydrate metabolic network
Generally, the prokaryote carbohydrate metabolic pathways are diverse, including the EMP pathway, ED pathway and PPP pathway. However, the genomic annotation revealed that the genes coding 6-phosphofructokinase (EC 2.7.1.11, Pfk), 2-keto-3-deoxy-6-phosphogluconate aldolase (EC 4.1.2.14, Eda) and phosphogluconate dehydratase (EC 4.2.1.12, Edd) were missing in strain SPU B805. Because Pfk is an enzyme specific to the EMP pathway, the absence of pfk leads to the incompleteness of the EMP route even though all the other enzymes are present. Similarly, Eda and Edd are the key enzymes unique to the ED pathway, therefore the absence of eda and edd imply the ED pathway is disabled in strain SPU B805. Thus, the PPP pathway was speculated as the main carbohydrate metabolic pathway in strain SPU B805.
The EMP pathways of strain WSH-001, Y25 and Hbe602 were also analyzed based on their published genome, and found to be incomplete because of the absence of gene pfk (Table 3), therefore, we speculated that the ED and PPP pathways were the main carbohydrate metabolic routes. However, for strain SKV, the gene pfk, eda and edd were all absent from the genome, so presumably the carbohydrate metabolism was mainly through the PPP pathway. To sum up, there are two types of carbohydrate metabolic pathways in K. vulgare. One is simultaneously through the ED and PPP pathways in strain WSH-001, Y25 and Hbe602, the other one is only through the PPP pathway in strain SKV and SPU B805. The loss of the ED pathway had no negative effective on 2-KGA production (Table 1) further confirmed that the ED pathway might be negligible in the cell growth and 2-KGA accumulation, thus the PPP pathway played an important role in the cytoplasmic carbohydrate metabolism in K. vulgare.
Carbohydrate metabolism analysis based on transcriptome
The abundance of FPKM (fragments per kilo bases of million fragments) was employed to represent the gene transcriptional level19. As for strain SPU B805, the whole FPKM was divided into 11 grades (from Rank −4 to Rank +6), and the median point was 195.564 (Fig. 4). The FPKM of the key genes related to PPP pathway and TCA cycle (Zwf, EC 1.1.1.49; Gnd, EC 1.1.1.44; Rpe, EC 5.1.3.1; RpiA, EC 5.3.1.6; TktA, EC 2.2.1.1; Tal, EC 2.2.1.2; GltA, EC 2.3.3.1; AceE, EC 1.2.4.1; Acn, EC 4.2.1.3; Icd, EC 1.1.1.42; SucA, EC 1.2.4.2; SucDC, EC 6.2.1.5; SdhA, EC 1.3.5.4; FumC, EC 4.2.1.2; Mdh, EC 1.1.1.37) were marked out in Fig. 4, and it was noteworthy that all of the genes related to carbohydrate metabolism (PPP, TCA) were transcribed at higher levels than the median point. However, no transcription signals of pfk, ga2dh, edd and eda could be detected, which confirmed that these genes did not exist in the genome of strain SPU B805, and indicated that the EMP and ED pathway were disabled. Because the PPP pathway, ED pathway and EMP pathway were three parallel carbon metabolic routes before TCA cycle, therefore, the only existing complete PPP pathway was assigned as the major and important cytoplasmic carbohydrate decomposition pathway linked with TCA cycle in strain SPU B805.

Transcriptome information of K. vulgare SPU B805. X axial represents the FPKM of K. vulgare SPU B805, Y axial represents the counts of expressed genes. The blue and pink panels indicate the FPKM is below and above the median point, respectively. The FPKM between 150 and 300, 300 and 600, 600 and 1200, 600 and 1200, 1200 and 2400, 2400 and 4800, 4800 and 9600 was defined as Rank 0, +1, +2, +3, +4, +5. The FPKM between 75 and 150, 36 and 75, 18 and 36 was defined as Rank −1, −2, and −3. The FPKM above 9600 and below 18 was defined as Rank +6 and Rank −4.
In fermentation, the FPKM of the five sdh (KVC_2764, KVC_2744, KVC_0718, KVC_1927, KVC_0337) (the average FPKM value 24691.32, 10501.89, 2078.73, 750.78, 220.99) were obviously higher than those of the central carbon degradation related genes, which partially explained that the majority of L-sorbose was converted to 2-KGA in strain SPU B805. Although the gene sndh (KVC_0605, the average FPKM value 479.86) had a relative lower transcriptional level, it did not affect the 2-KGA production rate because SDH was a dual-function enzyme, which could oxidize L-sorbose to L-sorbosone and further oxidize to 2-KGA20. Therefore, the central carbon metabolism, as well as the transcription level (the average FPKM value), were put together in Fig. 5. Strain SPU B805 possesses two separated modes for L-sorbose catabolism. A major part of L-sorbose is oxidized to L-sorbosone in the periplasm by the membrane-bound SDH, and further to 2-KGA by SDH and SNDH. The produced 2-KGA can’t be further decomposed to idonate because of the absence of plasmid 2 (so absence of gene ga2dh), therefore the 2-KGA production can reach a higher level in SPU B805 than that in Hbe602 and WSH-001. A minor part of L-sorbose as carbon source is catabolized to F6P and subsequently to 6PG in cytoplasm, and then enters the PPP pathway for energy and biomass production. In the oxidative branch, 6PG is converted into ribulose 5-phosphate (Ru5P), carbon dioxide and NADPH. NADPH is the major reducing power used to maintain the redox balance under stress situation21. The formed Ru5P is converted to R5P by ribose 5-phosphate isomerase (RpiA, EC 5,3,1,6) or X5P by ribulose 5-phosphate epimerase (Rpe, EC 5.1.3.1) in the non-oxidative branch. R5P and X5P undergo carbon rearrangement to generate sedoheptulose 7-phosphate (S7P), glyceraldehyde 3-phosphate (G3P) and erythorse 4-phosphate (E4P). R5P and E4P, as the precursors, were used for the biosynthesis of RNA, DNA, as well as aromatic amino acids13.

Overview of the average FPKM of carbohydrate metabolism related genes in K. vulgare SPU B805. Blue lines and orange lines indicate the average FPKM is lower and higher than the median point, respectively. Dotted lines indicate no transcription signal is detected. The thickness of lines represents the FPKM level. The figures above or below lines represent the EC number of the enzyme and the average FPKM.
RT-qPCR validation of the genes related to carbohydrate metabolism
The key genes in PPP pathway, zwf (KVC_1674), gnd (KVC_0890), transketolase (KVC_2247; tktA) and transaldolase B (KVC_1789; tal) were conducted by RT-qPCR. Compared with internal standard (polA), all of the four genes showed a relative high transcription level, and the changed trends in different time-points were in accordance with the transcriptome results (see Supplementary Fig. S6). The gene transcription level assayed by RNA-seq and RT-qPCR showed high correlation, indicating the reliability of the RNA-seq analysis.
Discussion
For a long time, researchers have mainly concentrated on elucidating the symbiosis mechanism of K. vulgare and B. megatherium, however, few studies have been explored on the underlying mechanism for the differences in central carbon metabolic pathways and 2-KGA yield of K. vulgare. In this study, the genome of K. vulgare SPU B805 was completely sequenced and the metabolic network was reconstructed, which provided a new insight into the carbohydrate metabolic specificity. K. vulgare SPU B805 was the first 2-KGA-producing strain without any plasmid. Its genome was similar to K. vulgare SKV except for some insertion, dislocation and rearrangement. The genome annotation showed that except for histidine, alanine and asparagine, the biosynthetic pathway of other amino acids was complete in strain SPU B805, which had significantly integral function than that in WSH-001 (eight amino acid biosynthesis pathways incomplete)6. Liu, et al.7 reported that glycine, proline, threonine and isoleucine play vital roles in K. vulgare growth and 2-KGA production, and the addition of these amino acids increased the 2-KGA productivity by 20.4%, 17.2%, 17.2% and 11.8%, respectively. Zhang, et al.8 also determined the key factors affected 2-KGA fermentation by orthogonal design experiments, and the results indicated that glycine and threonine were the key components affecting the mixed cell growth; serine, glycine, and proline were the key components that affected the 2-KGA production. Therefore, the more complete amino acid biosynthetic pathway, the more contribution to the high 2-KGA production of K. vulgare SPU B805. Moreover, the absence of the SNDH on plasmid 2 can avoid the unknown byproduct in the formation of 2-KGA. Besides, the gene ga2dh, responsible for the subsequent decomposition of 2-KGA to idonate, was also absent from strain SPU B805, which was a very advantageous factor for the 2-KGA accumulation. Additionally, the higher transcriptional level of five sdh (KVC_2764, KVC_2744, KVC_0718, KVC_1927, KVC_0337) helps to explain the high L-sorbose conversion ability of strain SPU B805.
The genome and transcriptome analysis showed that the central carbon metabolism of K. vulgare was versatile. Strain WSH-001, Y25 and Hbe602 contained all the genes involved in the ED, the PPP and the TCA cycle, meaning that these pathways were complete2,3,4. While the pfk gene was absent from strain WSH-001, Y25 and Hbe602 genome, indicating that the EMP pathway was nonfunctional in these strains. Whereas, in strain SPU B805 and SKV, all the genes referred to the PPP and the TCA cycle were found, meaning that the PPP and the TCA pathway were complete5. The pfk, as well as eda and edd genes were simultaneously missing from the genome of strain SPU B805 and SKV, which suggested that neither of the EMP and the ED pathway were functional. Therefore, the PPP pathway is the exclusively pathway for cytoplasmic carbohydrate decomposition in strain SPU B805 and SKV. In strain WSH-001, Y25 and Hbe602, the ED and the PPP pathways are paralleled for cytoplasmic carbohydrate catabolism. Although the ED pathway was complete in strain WSH-001, Y25 and Hbe602 genome, the carbon flux through this pathway is presumably negligible because of the lower ATP productivity ability of the ED pathway10. Moreover, in G. oxydans 621H, the deletion of gnd to inactive the PPP pathway resulted in a reduced final biomass and a decreased end-product 2-keto-gluconate, whereas, the inactivation of the ED pathway by deleting the edd-eda genes caused a lower overall sugar uptake by cells for the formation of biomass and energy in the cytoplasm, and led to a larger fraction of the sugar being converted to the final product of 2-keto-gluconate in the periplasm16. This research indicated that the PPP pathway was of the major importance for cytoplasmic carbohydrate catabolism, whereas the ED pathway is dispensable. The loss of ED pathway maybe has a similar effect on 2-KGA production in K. vulgare. Although higher ATP productivity in the EMP pathway than in the ED pathway, the EMP requires more enzymes to sustain the equivalent carbon flux10. If the protein synthesis is a limiting factor for cell growth, the bacterium usually does not synthesize additional protein only for one more energy22,23.
The PPP pathway, as a fundamental component in cellular metabolism, plays an important role in maintain carbon homoeostasis13. The PPP pathway can provide various intermediate metabolites for the biosynthesis of nucleotide and amino acids, such as G3P, E4P, R5P, S7P, and generate NADPH for the maintenance of redox balance under the stress situations21. K. vulgare as an aerobic bacterium, its growth and metabolism are inevitably subject to reactive oxygen species (ROS)24,25, therefore a reducing environment is an essential requirement for cell metabolism. It is well known that NADPH is the major reducing power in cell, and plays an important role in the GSH and thioredoxin metabolism26,27,28. Besides, a large number of anti-oxidant enzymes are coupled with NADPH as cofactor29, thus the PPP pathway may help K. vulgare SPU B805 to defeat intracellular ROS. Moreover, the biomass of K. vulgare was composed of protein, DNA, RNA, lipids, peptidoglycans, lipopolysaccharides, glycogen and soluble pool6,30. It is conceivable that the PPP pathway may provide a variety of intermediates as precursors to satisfy the demands for the biosynthesis of cellular components and facilitate to curb different types of environmental hardship in the process of growth.
Materials and Methods
Bacterial strains
Two strains, K. vulgare SPU B805 (2-KGA-producing strain) and B. megatherium SPU B806 (the helper strain), which were stored in Microbial Resource Center of Shenyang Pharmaceutical University, were used for 2-KGA fermentation in this study.
Culture medium and fermentation
The plate medium (P medium) contained 20 g/L L-sorbose, 1 g/L KH2PO4, 3 g/L yeast powder, 0.2 g/L MgSO4, 3 g/L beef extract, 1 g/L urea, 10 g/L peptone, 3 g/L corn steep liquor and 20 g/L agar, pH 6.8. The plates were cultured at 28–30 °C for 4 days. The seed medium (S medium) contained 20 g/L L-sorbose, 1 g/L KH2PO4, 3 g/L yeast powder, 0.2 g/L MgSO4, 12 g/L urea, 10 g/L corn steep liquor, pH 6.8. Seeds were cultured at 28–30 °C, 250 rpm for 18 h. The fermentation medium (F medium) contained 80 g/L L-sorbose, 20 g/L corn steep liquor, 1 g/L KH2PO4, 0.2 g/L MgSO4, 12 g/L urea. The fermentation was conducted in 5-L fermenter containing 3 L F medium and 300 mL co-inoculated seeds of K. vulgare SPU B805 and B. megatherium SPU B806. The fermentation pH was maintained automatically at 6.8–7.0 by addition of 30% (m/v) Na2CO3 solution. The temperature was controlled at 30 °C, and the agitation speed was 400 rpm with the air flow rate of 1.5 L/min.
To calculate the Colony Forming Units (CFUs) of K. vulgare, the fermentation broth was diluted and spread on P plate, and the growth curve was drawn according to the CFU/mL. Meanwhile, the cell density was measured spectrophotometrically at 660 nm.
Genome sequencing and assembly
K. vulgare SPU B805 was cultured in 250 mL flasks with 50 mL S medium at 30 °C for 18 h at 220 rpm. The genome of strain SPU B805 was extracted with the TIANamp Genomic DNA Kit (Tiangen Biotech (Beijing) Co., Ltd.) according to the instruction. The concentration and purity of DNA sample were qualified by Agilent 2100 BioAnalyzer (Agilent Technologies, USA). The integrity of genomic DNA was detected by agarose gel electrophoresis. The DNA samples with a 260/280 nm absorbance ration of 1.8–2.0 and a 260/230 nm absorbance ration of 2.0–2.2 were considered pure and then used for the library construction and sequencing.
The complete genome sequence was determined by the Chinese National Human Genome Center (Shanghai, China) using 454 single-end sequencing technology. It yielded a total of 116,450 reads with a 17.29-fold coverage of the genome. The reads were assembled into 26 large contigs (>500 nucleotides) and 3 small contigs (100–500 nucleotides) by using the 454 Newbler assembler (454 Life Sciences, Branford, CT). The relationships between the contigs were determined by ContigScape31, and the gaps between the contigs were closed by PCR amplification, followed by DNA sequencing. Finally, the contigs were assembled into a circular chromosome, and no plasmid was found. The complete genome of SPU B805 had an error rate of less than 0.2 in a 10-kb sequence as determined by sequence assembly and quality assessment using the Phred32/Phrap33/Consed34 software package.
Genome annotation
Protein-coding genes were predicted by combining the results of Glimmer 3.0235 and ZCURVE36, followed by manual inspection. Each gene was functionally classified by assigning a Cluster of Orthologous groups (COG) number. Both tRNA and rRNA genes were identified by tRNA scan-SE37 and RNAmmer38, respectively. Functional annotations of ORFs were performed by searching against public databases, including NCBI (http://www.ncbi.nlm.nih.gov/), KEGG39 and SWISS-Prot40. Basic metabolic pathways were reconstructed by KEGG and MetaCyc41. The Rapid Annotation using Subsystem Technology (RAST) server42 was used to re-annotate the genome of K. vulgare SPU B805. Then, RAST annotation was put through the Model SEED pipeline to obtain the initial network reconstruction43.
Transcriptome sequencing
K. vulgare SPU B805 and B. megatherium SPU B806 were co-cultured in 5-L fermenter. The fermentation processes of 0 h (seed), 8 h, 16 h, 24 h, 32 h and 40 h were chosen to represent different growth phase. The co-culture samples were subjected to a centrifugation of 4,000 rpm for 5 min at 4 °C to eliminate the cells of B. megatherium SPU B806. The RNA of K. vulgare SPU B805 was extracted with the RNAprep Pure Bacteria Kit (Tiangen, Beijing, China). The concentration and purity of RNA sample were qualified by a Agilent 2100 BioAnalyzer (Agilent Technologies, USA). The integrity was monitored on 1% agarose gels. The RNA samples with a 260/280 nm absorbance ration of 1.8–2.1 were considered pure, and a rRNA ration (28S/18S) ≥1.5 and RIN ≥7 were considered integrity. The NEBNext Ultra RNA Library Prep Kit (NEB, USA) for Illumina was used to construct sequencing libraries. The rRNA was removed from total RNA using a Ribo-Zero Magnetia Kit (Epicentre, USA) to enrich the mRNA and then the cDNA was synthesized. The library preparation was sequenced on Illumina Hiseq 2500 platform. The genome of K. vulgare SPU B805 was used as a reference.
Analyses of 2-KGA and L-sorbose
The concentration of 2-KGA and L-sorbose in the fermentation broth were analyzed by the High-Performance Liquid Chromatography (HPLC) (Waters Corp., Massachusetts, USA) using an amino column (Agilent Zorbax NH2) with an ultraviolet detector at 210 nm. Cetonitrile-KH2PO4 (5%/95%, v/v) was used as mobile phase with a flow rate of 0.6 mL/min.
Real time quantitative PCR
The primers used for real time quantitative PCR (RT-qPCR) were listed in Supplementary Table S3. RT-qPCR was conducted with the GoTaq qPCR Master Mix (Promega (Beijing) Biotech Co., Ltd) on a Stratagene Mx3000P (Agilent Technologies Inc, USA). The DNA polymerase I (KVC_2082, polA) gene was used as an internal standard. The fold change of gene expression was calculated by the 2−ΔΔCT method44.
References
Zhang, J., Liu, J., Shi, Z., Liu, L. & Chen, J. Manipulation of B. megaterium growth for efficient 2-KLG production by K. vulgare. Process Biochem. 45, 602–606 (2010).
Xiong, X. H. et al. Complete genome sequence of the bacterium Ketogulonicigenium vulgare Y25. J. Bacteriol. 193, 315–316 (2011).
Liu, L. et al. Complete genome sequence of the industrial strain Ketogulonicigenium vulgare WSH-001. J. Bacteriol. 193, 6108–6109 (2011).
Jia, N. et al. Insights into mutualism mechanism and versatile metabolism of Ketogulonicigenium vulgare Hbe602 based on comparative genomics and metabolomics studies. Sci. Rep. 6, 23068 (2016).
Jia, N. et al. Complete Genome Sequence of the Industrial Bacterium Ketogulonicigenium vulgare SKV. Genome Announc 4 (2016).
Zou, W. et al. Reconstruction and analysis of a genome-scale metabolic model of the vitamin C producing industrial strain Ketogulonicigenium vulgare WSH-001. J. Biotechnol. 161, 42–48 (2012).
Liu, L., Chen, K., Zhang, J., Liu, J. & Chen, J. Gelatin enhances 2-keto-L-gulonic acid production based on Ketogulonigenium vulgare genome annotation. J. Biotechnol. 156, 182–187 (2011).
Zhang, J. et al. Development of chemically defined media supporting high cell density growth of Ketogulonicigenium vulgare and Bacillus megaterium. Bioresource Technol. 102, 4807–4814 (2011).
Fuhrer, T., Fischer, E. & Sauer, U. Experimental identification and quantification of glucose metabolism in seven bacterial species. J. Bacteriol. 187, 1581–1590 (2005).
Flamholz, A., Noor, E., Bar-Even, A., Liebermeister, W. & Milo, R. Glycolytic strategy as a tradeoff between energy yield and protein cost. PNAS. 110, 10039–10044 (2013).
Chen, X. et al. The Entner-Doudoroff pathway is an overlooked glycolytic route in cyanobacteria and plants. PNAS. 113, 5441–5446 (2016).
Siebers, B. & Schonheit, P. Unusual pathways and enzymes of central carbohydrate metabolism in Archaea. Curr. Opin. Microbiol. 8, 695–705 (2005).
Stincone, A. et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 90, 927–963 (2015).
Du, J., Bai, W., Song, H. & Yuan, Y. J. Combinational expression of sorbose/sorbosone dehydrogenases and cofactor pyrroloquinoline quinone increases 2-keto-L-gulonic acid production in Ketogulonigenium vulgare-Bacillus cereus consortium. Metab. Eng. 19, 50–56 (2013).
Chen, S., Jia, N., Ding, M. & Yuan, Y. Fitness analysis between the L-sorbosone dehydrogenase modules and Ketogulonigenium vulgare chassis. Chin. J. Biotechnol. 32, 1224–1232 (2016).
Richhardt, J., Bringer, S. & Bott, M. Role of the pentose phosphate pathway and the Entner–Doudoroff pathway in glucose metabolism of Gluconobacter oxydans 621H. Appl. Microbiol. Biot. 97, 4315–4323 (2013).
Wang, C. Y. et al. Establishing an innovate carbohydrate metabolic pathway for efficient production of 2-keto-L-gulonic acid in Ketogulonicigenium robustum initiated by intronic promoters. Microb. Cell Fact. 17, 81 (2018).
Takagi, H. Proline as a stress protectant in yeast: physiological functions, metabolic regulations, and biotechnological applications. Appl. Microbiol. Biot. 81, 211–223 (2008).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Asakura, A. & Hoshino, T. Isolation and characterization of a new quinoprotein dehydrogenase, l-sorbose/l-sorbosone dehydrogenase. Biosci. Biotechnol. Biochem. 63, 46–53 (1999).
Kruger, A. et al. The pentose phosphate pathway is a metabolic redox sensor and regulates transcription during the antioxidant response. Antioxid. Redox. Sign. 15, 311–324 (2011).
Molenaar, D., van Berlo, R., de Ridder, D. & Teusink, B. Shifts in growth strategies reflect tradeoffs in cellular economics. Mol. Syst. Biol. 5, 323 (2009).
Scott, M., Gunderson, C. W., Mateescu, E. M., Zhang, Z. & Hwa, T. Interdependence of cell growth and gene expression: origins and consequences. science 330, 1099–1102 (2010).
Ma, Q. et al. Proteomic analysis of Ketogulonicigenium vulgare under glutathione reveals high demand for thiamin transport and antioxidant protection. Plos one 7, e32156 (2012).
Ma, Q. et al. Integrated proteomic and metabolomic analysis of an artificial microbial community for two-step production of vitamin C. Plos one 6, e26108 (2011).
Lushchak, V. I. Oxidative stress and mechanisms of protection against it in bacteria. Biochemistry 66, 476–489 (2001).
Pandolfi, P. P. et al. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. Embo J. 14, 5209–5215 (1995).
Prinz, W. A., Aslund, F., Holmgren, A. & Beckwith, J. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J. Biol. Chem. 272, 15661–15667 (1997).
Cabiscol, E., Tamarit, J. & Ros, J. Oxidative stress in bacteria and protein damage by reactive oxygen species. Int. Microbiol. 3, 3–8 (2000).
Feist, A. M. et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Sys. Biol. 3, 121 (2007).
Tang, B. et al. ContigScape: a Cytoscape plugin facilitating microbial genome gap closing. Bmc Genomics 14 (2013).
Ewing, B., Hillier, L. D., Wendl, M. C. & Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy Assessment. Genome Res. 8, 175–185 (1998).
de la Bastide, M. & Mccombie, W. R. Assembling genomic DNA sequences with PHRAP. Curr. Protoc. Bioinformatics Chapter 11, Unit11.14 (2007).
Gordon, D. & Green, P. Consed: a graphical editor for next-generation sequencing. Bioinformatics 29, 2936–2937 (2013).
Delcher, A. L., Bratke, K. A., Powers, E. C. & Salzberg, S. L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679 (2007).
Guo, F. B., Ou, H. Y. & Zhang, C. T. ZCURVE: a new system for recognizing protein-coding genes in bacterial and archaeal genomes. Nucleic Acids Res. 31, 1780–1789 (2003).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Kanehisa, M. et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34, D354–357 (2006).
Boeckmann, B. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 (2003).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 42, 459–471 (2014).
Overbeek, R. et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–214 (2014).
Henry, C. S. et al. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 28, 977–982 (2010).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 25, 402–408 (2001).
Acknowledgements
This work was supported by grant from the national science and technology project (31370125, 81703397, 2018ZX09735001-002-002) and Liaoning science and technology project (LR2015065, 2017LQN17, 2017107013).
Author information
Authors and Affiliations
Contributions
C.Y.W., Y.L. designed the project and experiments; C.Y.W. and Z.W.G. performed the experiments; Y.L. and L.C.L. annotated and uploaded the genome; Y.C.W. assisted in the analysis of final data. M.Y.Z. and T.Y.Z. assisted in the revision the manuscript; Y.X.Z. revised the manuscript. All the authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, CY., Li, Y., Gao, ZW. et al. Reconstruction and analysis of carbon metabolic pathway of Ketogulonicigenium vulgare SPU B805 by genome and transcriptome. Sci Rep 8, 17838 (2018). https://doi.org/10.1038/s41598-018-36038-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36038-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
