
Abstract
As an important transcription factor, SOX2 involves in embryogenesis, maintenance of stem cells and proliferation of primordial germ cell (PGC). However, little was known about its function in mature gonads. Herein, we investigated the SOX2 gene profiles in testis of scallop, Chlamys farreri. The level of C. farreri SOX2 (Cf-SOX2) mRNA increased gradually along with gonadal development and reached the peak at mature stage, and was located in all germ cells, including spermatogonia, spermatocytes, spermatids and spermatozoa. Knockdown of Cf-SOX2 using RNAi leaded to a mass of germ cells lost, and only a few spermatogonia retained in the nearly empty testicular acini after 21 days. TUNEL assay showed that apoptosis occurred in spermatocytes. Furthermore, transcriptome profiles of the testes were compared between Cf-SOX2 knockdown and normal scallops, 131,340 unigenes were obtained and 2,067 differential expression genes (DEGs) were identified. GO and KEGG analysis showed that most DEGs were related to cell apoptosis (casp2, casp3, casp8), cell proliferation (samd9, crebzf, iqsec1) and spermatogenesis (htt, tusc3, zmynd10, nipbl, mfge8), and enriched in p53, TNF and apoptosis pathways. Our study revealed Cf-SOX2 is essential in spermatogenesis and testis development of C. farreri and provided important clues for better understanding of SOX2 regulatory mechanisms in bivalve testis.
Similar content being viewed by others


Introduction
SOX2 is a member of the SOX (SRY-related HMG-box) family with a high-mobility-group (HMG) DNA-binding domain. Several studies have determined that SOX2, as an important transcription factor, participates in development processes of vertebrates, such as embryogenesis1,2, maintenance of stem cells3,4,5, neurogenesis6,7,8 etc. Recent investigations have also emerged that SOX2 is one of the transcription factors underlying the derivation of induced pluripotent stem cells9,10,11.
The role of SOX2 in gametogenesis has only been reported in mouse Mus musculus. Campolo et al.12 found that SOX2 participates in the proliferation of primordial germ cells (PGCs). However, the role of SOX2 in gametogenesis of mature adult of vertebrates and invertebrates has not been reported. Up to now, studies about SOX2 related to gonad development are limited. Researchers found that SOX2 mRNA is ubiquitously expressed in all tissues in Hyriopsis schlegelii and the expression level in the testis is higher than that in ovary13; besides, SOX2 expression in testis raised gradually along with its development14, which could be divided into three stages: initiation of testis formation (4–11 months), a stable growth phase (12–24 months), and a reproductive cell development phase (25–36 months), indicating its role in the development of testis13. In Paralichthys olivaceus, SOX2 transcripts is expressed in gonadal tissues and the transcript abundance in ovary is higher than that in testis15. Patra et al.16 found that, in rohu carp Labeo rohita, SOX2 mRNA is expressed in various organs, as well as in the cultured proliferating spermatogonial stem cells (SSCs). The SOX2 expression level is highest in SSCs and its transcript abundance is higher in testis than that in ovary. Moreover, SOX2 protein is presented in SSCs of L. rohita, suggesting the participation of SOX2 in male germ cell development and stem cells maintenance of fish16.
Spermatogenesis is a complex process and lots of genes have been discovered involving in this process, such as stra8, nanos3, ddx5, tspy1, ngn3, etc.17,18. However, most of these researches were performed in vertebrates or model organisms, while in mollusks, the related studies are limited. As for bivalves, several spermatogenesis related genes (adad1, shippo1, etc.) were screened out through pyrosequencing in giant lion’s paw Nodipecten subnodosus19. In Zhikong scallop Chlamys farreri, multiple genes have been cloned and proposed playing roles in gametogenesis based on their expression pattern (DAX1, β-Catenin and FOXL2)20,21,22, while recently KLF4 and PIWI1 genes were validated involving in spermatogenesis and gametogenesis, respectively, through RNAi23,24. Besides, function of many sox family genes in gametogenesis have been identified and studied, such as SOX2, SOX7, SOX 8, SOX 14 in pacific oyster Crassostrea gigas25; SOX9 in pearl oyster Pinctada margaritifera26; SOXE, SOXH in Yesso scallop Patinopecten yessoensis27; SOX2 and SOXB2 in C. farreri28,29 and SOX11 in Pinctada martensii30. However, the studies of these SOX genes are confined to the expression analysis. In this study, Zhikong scallop C. farreri, an important commercial marine bivalve in China, was employed and the abundance and cyto-location of C. farreri SOX2 (Cf-SOX2) mRNA in testis were revealed by qRT-PCR and in situ hybridization. We further determined that Cf-SOX2 participated in the regulation of C. farreri spermatogenesis and testis development by means of RNA interference (RNAi). Moreover, we performed transcriptome analysis on testes before and after Cf-SOX2 knockdown and investigated expression profiles of SOX2-related genes. Our data provide important clues for better understanding of the molecular mechanism about spermatogenesis and testis development in bivalve mollusks.
Results
Temporal expression of Cf-SOX2 in testes during the reproductive cycle
The relative levels of Cf-SOX2 mRNA in testes were examined by qRT-PCR (Fig. 1). Abundance of the Cf-SOX2 transcript increased significantly (P < 0.05) from proliferative stage to mature stage. And the expression levels were 2-fold and 4-fold higher in growing stage and mature stage than that in proliferative stage, respectively.

Relative expression levels of Cf-SOX2 mRNA in C. farreri testes detected by qRT-PCR. Values are the mean ± SEM; n = 5. The expression level in the testis at proliferative stage is set as 1.00 to calibrate the relative expression at other stages. Different letters (a–c) indicate statistically significant differences (P < 0.05).
Cyto-location of Cf-SOX2 mRNA in testis during spermatogenesis
In C. farreri testis, spermatogonia, spermatocytes, spermatids and spermatozoa successively arranged from follicle wall to central region of germinal acini. The spatial location of Cf-SOX2 mRNA in testes at different stages was demonstrated using in situ hybridization (Fig. 2). The Cf-SOX2 transcripts were located in all germ cells, while intensity of the positive hybridization signals in spermatocytes was slightly higher than that in spermatogonia, spermatids and spermatozoa (Fig. 2a,b,d–f,g–h). Moreover, no hybridization signal was detected in testes with the sense probe (Fig. 2c,i).

Cyto-location of Cf-SOX2 mRNA in testes of C. farreri during spermatogenesis demonstrated by in situ hybridization. Positive signals with an antisense probe are indicated in blue (a,b and d–h). The negative control is conducted with sense probe (c and i). (a–c), Proliferative stage; (d–e), growing stage; (f–i), mature stage. Sg, spermatogonium; Sc, spermatocyte; St, spermatid; Sz, spermatozoon. Scale bar, 25 μm for h and 50 μm for others.
SOX2 knockdown led to a testis developmental retardance in C. farreri
The efficiency of Cf-SOX2 knockdown was examined by qRT-PCR (Fig. 3). The level of Cf-SOX2 mRNA in the SOX2-dsRNA testes was significantly decreased (P < 0.05), which was only 35% of that in the blank group. No significant difference of Cf-SOX2 mRNA level was detected between the blank group and EGFP-dsRNA group.

Level of Cf-SOX2 mRNA in C. farreri testes on 72 h post injection of RNAi detected by qRT-PCR. Values are the mean ± SEM, n = 5; The expression level in the testes of blank group is set as 1.00 to calibrate the relative expression of other groups. Different letters (a,b) indicate statistically significant differences (P < 0.05).
The phenotype of the scallops presented obvious differences between the SOX2-dsRNA group and the control groups after Cf-SOX2 mRNA was knocked down. In the SOX2-dsRNA injected group, the testes were translucent and wizened, and their size was much smaller than that of the control groups (Fig. 4a). The gonadosomatic index (GSI) of the SOX2-dsRNA injected scallops (3.68 ± 0.82) was significantly (P < 0.05) lower than that of the EGFP-dsRNA injected scallop (6.07 ± 0.52) or the blank scallops (6.45 ± 0.81) (Fig. 4b).

Characteristics of C. farreri testes after Cf-SOX2 knockdown. (a) external morphology of C. farreri testes on the 21st day post RNAi; (b) GSI of C. farreri on the 21st day post RNAi (n = 6); (c) gametes number in testes of a 6400 μm2 area on the 3rd day post RNAi (n = 3, three sights of each sample, respectively); (d) percentage of various types of germ cells in a 6400 μm2 area on the 3rd day post RNAi (n = 3, three sights for each sample, respectively). Data are means ± SEM. Different letters indicate significant differences (P < 0.05).
The knockdown of Cf-SOX2 expression level provoked defects of the germ cell development in testis (Fig. 5). On the 3rd day post injection, the germ cells in SOX2-dsRNA group loosely arranged in the germinal acini (Fig. 5c), and the number of the germ cells (70.0 ± 19.3 in 6400 μm2) reduced about 68% compared with the blank testis (223.0 ± 8.3 in 6400 μm2) which had no significant difference from that in EGFP-dsRNA testis (215.5 ± 19.6 in 6400 μm2) (Fig. 4c). Furthermore, cells in testes were labeled with Cf-SOX2, as well as the C. farreri germ cell marker gene VASA (Cf-VASA), through in situ hybridization, in order to identify different types of germ cells. The Cf-VASA transcripts were located in spermatogonia, spermatocytes, spermatids (Fig. 6a–c). While, in SOX2-dsRNA group, Cf-SOX2 mRNA were weakly expressed in spermatogonia and few spermatocytes (Fig. 6g). No hybridization signal was detected in testes with the sense probe (Fig. 6d,h). On the 10th day post injection, only a few number of spermatogonia and spermatocytes loosely arranged in the germinal acini in SOX2-dsRNA group, while the germinal acini in control groups were filled with all kinds of germ cells (spermatogonia, spermatocytes, spermatids, spermatozoa) (Fig. 5d–f). When it is on the 21st day post injection, the germinal acini were nearly empty, and only several spermatogonia were observed in SOX2-dsRNA group (Fig. 5i). Moreover, the composition of germ cells was also changed on the 3rd day post injection. In SOX2-dsRNA testes, the germ cell composition was 20.8% spermatogonia, 47.2% spermatocytes, 18% spermatids and 14% spermatozoa, comparing to 8.0%, 59.2%, 20% and 21.1% of that in blank group which was similar in EGFP-dsRNA testes (Fig. 4d).

Histological observation of the C. farreri testes after the knockdown of Cf-SOX2. (a–c) Testes on the 3rd day post RNAi, the blank group (a), the EGFP-dsRNA group (b) and the SOX2-dsRNA group (c); (d–f), testes on the 10th day post RNAi, the blank group (d), the EGFP-dsRNA group (e) and the SOX2-dsRNA group (f); (g–i), testes on the 21st day post RNAi, the blank group (g), the EGFP-dsRNA group (h) and the SOX2-dsRNA group (i). Sg, spermatogonium; Sc, spermatocyte; St, spermatid; Sz, spermatozoon. Scale bar, 50 μm.

Status of germ cells in the C. farreri testes of 3 days post Cf-SOX2 knockdown demonstrated by in situ hybridization. (a–d) Cyto-location of Cf-VASA mRNA; (e–h) cyto-location of Cf-SOX2 mRNA. Positive signals with an antisense probe are indicated in blue (a–c,e–g). The negative control is conducted with sense probe (d,h). (a,e) The blank group; (h,f) the EGFP-dsRNA group; c,g the SOX2-dsRNA group. Sg, spermatogonium; Sc, spermatocyte; St, spermatid; Sz, spermatozoon. Scale bar, 10 μm.
TUNEL detection revealed that many spermatocytes in SOX2-dsRNA testes after RNAi for 3 days presented obvious positive signals of apoptosis, while no obvious signals were visible in spermatogonia, spermatids and spermatozoa (Fig. 7g,h). Besides, the obvious positive signals were invisible in testes of EGFP-dsRNA group and the blank group (Fig. 7a,b,d,e).

Cell apoptosis analysis by TUNEL in the C. farreri testes of 3 days post Cf-SOX2 knockdown. Positive signals are indicated in brown. (a–c) The blank group; (d–f) the EGFP-dsRNA group; (g–i), the SOX2-dsRNA group; (b,e,h) magnified images of the gonads; (c,f,i) negative control without terminal deoxynucleotidyl transferase. Sg, spermatogonium; Sc, spermatocyte; St, spermatid; Sz, spermatozoon. Scale bar, 10 μm.
Transcriptome sequencing and de novo assembly
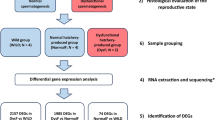
Transcriptome analysis were conducted from six testes of C. farreri in SOX2-dsRNA and blank groups (Table S1) and a total of 272,353,746 raw reads were generated (Table S2). After removing the adaptor sequences and ambiguous or low-quality reads, we obtained a total of 263,383,562 clean reads (96.70% of the raw reads), generating 131,340 assembled unigenes which had an average length of 1,149 bp and an N50 of 1,807 bp (Table S2, Fig. S1). All data have been submitted to the NCBI Sequence Read Archive (SRP125788).
Functional annotation
Among the 131, 340 unigenes detected in the C. farreri testis, 56,396 (42.9%) were successfully annotated with at least one database (Table S2). According to the GO annotation, 41,327 unigenes were assigned to 55 level-2 GO terms under the three main GO categories, biological process, cellular component and molecular function (Fig. 8a). Cellular process (23441, 56.7%), metabolic process (19791, 47.9%) and single-organism process (18526, 44.8%) were the three most highly represented GO terms within the biological process, whereas binding (23129, 56.0%) and catalytic activity (15616, 37.8%) were the two most represented terms in molecular function.

Function annotation of all unigenes in C. farreri testis. (a) Histogram presentation of the Gene Ontology (GO) classification; (b) Histogram presentation of clusters of eukaryotic Ortholog Groups (KOG) classification; (c) Histogram presentation of Kyoto Encyclopedia of Genes and Genomes (KEGG) classification. A, Cellular Processes; B, Environmental Information Processing; C, Genetic Information Processing; D, Metabolism; E, Organismal Systems.
Orthologous gene products were classified with the KOG database. 17,186 unigenes were allocated to 26 KOG classifications (Fig. 8b), among which “general function prediction only” (2820, 16.4%) was the largest group, followed by “signal transduction mechanisms” (2711, 15.8%) and “posttranslational modification, protein turnover, chaperones” (1816, 10.6%).
KEGG analysis assigned 15,901 unigenes to specific categories (Fig. 8c). Most of them fell into “organismal system” (5037, 31.7%), followed by “metabolism” (4548, 28.6%), “cellular processes” (2756, 17.3%), “environmental information processing” (2571, 16.2%) and “genetic information processing” (2538, 16.0%). The top five KEGG pathway were “signal transduction” (2135, 13.4%), “endocrine system” (1274, 8.0%), “transport and catabolism” (1149, 7.2%), “translation” (932, 5.9%) and “folding, sorting and degradation” (882, 5.5%).
Differential expression analysis
A total of 2,067 DEGs were obtained between the SOX2-dsRNA group and bland group with FDR < 0.05 (Table S3), of which 993 (48.0%) were up-regulated and 1,074 (52.0%) were down-regulated after the knockdown of Cf-SOX2 (Fig. 9a). Clustering analysis showed that the expression patterns of DEGs were obviously different between the blank group and SOX2-dsRNA group (Figs. 9b), and 6 subclusters could be generated (Fig. S2).

Analysis of differential expressed genes in the testes of C. farreri between SOX2-dsRNA group and blank group. (a) The volcano plot of DGEs. Red spots indicate significantly up-regulated genes; green spots indicate significantly down-regulated genes; blue spots are genes of no different expression. The significant difference is set at FDR < 0.05 to identify the different expression genes between two groups; (b) Clustering analysis of DEGs. Each column represents a group, and each row represents a gene. Red represents high level of expression, and blue represents low level of expression; numbers represent expression abundance. C, blank group; E, SOX2-dsRNA group.
Based on GO annotation, a total of 38 DEGs were identified to involve in apoptosis, cell proliferation and reproduction (Table S3), such as regulation of apoptotic process (casp2, casp3, casp8), cell proliferation and growth (bmp7, samd9, crebzf, iqsec1), reproductive (htt, tusc3), mitotic cell cycle, meiotic cell cycle (tle4, rprd2) and spermatogenesis (zmynd10, nipbl, mfge8, zfand3 and armc3).
KEGG pathway enrichment analysis showed that DEGs were enriched in 180 pathways. Among these pathways, 5 pathways enriched more than 5 DEGs, including cytosolic DNA-sensing pathway (6), spliceosome (10), viral carcinogenesis (9), herpes simplex infection (7) and influenza A (7). Moreover, the DEGs related to apoptosis and anti-apoptosis genes, such as casp3, casp8, birc3 were enriched in apoptosis pathway, p53 signaling pathway and TNF signaling pathway (Table S4).
Quantitative RT-PCR validation
The expression profiles of genes identified in RNA-Seq analysis were confirmed by detecting the relative mRNA levels of the following 8 genes by qRT-PCR: casp2, casp3, casp8, crebzf, htt, tusc3, bmp7, ddx6 (Fig. 10). The results indicated that the data from qRT-PCR were basically consistent with those of RNA-Seq, and although the crebzf gene was not statistically significantly different in the qRT-PCR assay, it still showed the same differential expression tendency as revealed by RNA-Seq. The overall consistency confirmed the reliability of RNA-Seq data to accurately quantify the gene expression.

Validation of the RNA-Seq data of C. farreri testes using qRT-PCR. Vertical bars represent the qRT-PCR results; lines represent the FPKM values obtained from RNA-Seq analysis. Values are the mean ± SEM, n = 3; The expression level in the testes of blank group is set as 1.00 to calibrate the relative expression. Star (*) indicate statistically significant differences (P < 0.05).
Discussion
It has been reported that SOX2 gene participates in the proliferation of PGCs and other processes during embryo development2,5,11,12. However, the studies of SOX2 in mature gonad, especially in testis, are still limited, thus its function or participant biological processes are largely unclear. In this study, Cf-sox2 mRNA was expressed significantly and located in all germ cells during reproductive cycle of testis. The joint study of experimental and bioinformatics analysis revealed that Cf-SOX2 gene is essential to spermatogenesis and functional maintain in adult C. farreri testis. Our results will assist in better understanding of the regulatory mechanisms of SOX2 gene in bivalve testis.
Cf-SOX2 regulates proliferation and apoptosis of spermatogonia and spermatocytes in C. farreri
In M. musculus, SOX2 protein are expressed in primordial germ cells (PGCs) of mouse embryo at 7.5 days post coitum (dpc), and involves in the proliferation of PGCs and the formation of spermatogonia. Campolo et al.12 found that in mouse embryos with SOX2 conditionally knocked out by Cre/loxP, the number of PGCs is strongly reduced and spermatogonia in testes are completely depleted at 17.5 dpc. TUNEL and BrdU incorporation experiments show that the reduction of PGCs is mainly due to the decreased DNA synthesis rather than the increased apoptosis. Furthermore, proliferation of the cultured PGCs have also been verified to be associated with SOX2, with the fact that normal PGCs isolated from mouse embryo 8.5–11.5 dpc can proliferate in vitro, while those from the SOX2-knockout embryo do not.
In this study, the Cf-SOX2 transcripts were revealed to locate in all germ cells of C. farrier testis, although its abundance in spermatocytes was slightly higher than that in spermatogonia, spermatids and spermatozoa (Fig. 2a,b,d–h). Moreover, the number of all spermatogenic cells dramatically reduced (Fig. 4c, 5a-c, 6) in testes 3 days post the knockdown of Cf-SOX2, and then only few spermatogonia were observable (Fig. 5g–i) until the 21st day. Therefore, we suggested that SOX2 is involved in animal spermatogenesis besides regulating the proliferation of PGCs.
To explore the reason for the reduction of the spermatogenic cells, we carried out the TUNEL assay and found that obvious apoptosis was presented in spermatocytes of the SOX2-dsRNA testes (Fig. 7g,h). The differences between what we found in scallop and that in mouse implied SOX2 may involve in different mechanisms of regulating the differentiation of spermatogenic cells at different stages. Furthermore, we found the number of spermatogonia in SOX2-dsRNA testes declined gradually with the progress of RNAi (Fig. 5c,f,i), while no TUNEL positive signal was observed. Based on the fact above, we proposed that the significant reduction of germ cell number might be resulted from the apoptosis of spermatocytes as well as the disability of spermatogonia proliferation and differentiation, and then lead to the failure of spermatid and spermatozoon formation.
Cf-SOX2 regulates spermatogenesis and testis functional maintenance through several biological pathways in C. farreri
Based on the transcriptome analysis performed with the testes 3 days post RNAi, a total of 2,067 DEGs were obtained between Cf-SOX2 knockdown testis and normal testis. Functional and enrichment analysis showed that lots of DEGs and pathways have involved into the spermatogenesis and testis functional maintenance.
Some DEGs (samd9, crebzf, iqsec1, bmp7) involved in cell proliferation, mitosis and meiosis presented differential expression after Cf-SOX2 knockdown. samd9 is found to be related to cell proliferation and apoptosis. Its over expression in the colon cell line reduced the cell proliferation and boosted the CASP3 activity, while the proliferation rate restored after its knockdown with RNAi31. Analogous, the up-regulation of crebzf and iqsec1 result in the inhibition of cell proliferation in mouse and human cells, and the down-regulation of bmp7 result in the weaken of the proliferation ability32,33,34,35. In the present study, along with the testis retardance after Cf-SOX2 knockdown, these four genes presented either up- or down-regulated expression patterns and based on their coincidence with the reported studies, we speculated Cf-SOX2 could regulate the spermatogenesis through control the proliferation of germ cells.
Cell apoptosis is a process of programmed cell death, which controlled by a series of genes involving in the activation, expression and regulation processes. As the members of CASP family, CASP2 and CASP8 are apoptotic promoters, and CASP3 is the apoptotic executor, which plays an important role in cell apoptosis process36,37,38,39,40,41. cIAP2, encoded by birc3, is one of the members of IAPs (inhibitor of apoptotic proteins) family, which can block the apoptosis process by inhibiting the enzymatic activity of CASP3, CASP7 and CASP942,43,44. In this study, a variety of genes (casp2, casp3 and casp8) involved in cell apoptosis were up-regulated in testes after RNAi. Taking the results of TUNEL detection into consideration (Fig. 7g,h), we suggested that the activation of CASP apoptosis pathway in cells might be one of the reasons that resulted in the significant reduction of germ cells. Interestingly, the anti-apoptosis gene (birc3) was also up-regulated on the 3rd day of RNAi. We speculated it might be a stress response to cell apoptosis at the early stage.
Spermatogenesis is a process involving cellular proliferation, meiosis, and terminal differentiation that occurs during development of the testis and lots of specific genes involve in this process. zmynd10 and armc3 are important genes playing roles in sperm motility, and their deficiency cause sterility due to immotile sperm45,46,47,48. NIPBL is required for the association of cohesion with DNA and thus proper chromosome segregation. It is an important factor for mammals during early stages of meiotic prophase49. SED1, also known as MFG-E8, is expressed on the surface of sperm plasma membrane overlying the intact acrosome in mice and human, and plays an important role in sperm-egg binding50,51,52,53,54, while tusc3 is expressed in spermatocytes55, and they have been suggested to be involved in male germ cell maturation. Htt mRNA is located in both primary and secondary spermatocytes, and the knockout of htt leads to significant decrease of spermatocytes and spermatids56,57. In the present study, all these genes were down-regulated after the knockdown of Cf-SOX2, which indicated Cf-SOX2 could regulate the spermatogenesis process through these potential important downstream genes.
According to the KEGG pathway enrichment analysis, some pathways related to apoptosis, such as p53 signaling pathway, TNF signaling pathway and Apoptosis pathway, were enriched. P53 is a tumor suppressor protein that regulates the expression of a wide variety of genes involved in apoptosis, growth arrest, inhibition of cell cycle progression58. Tumor necrosis factor (TNF) also known as TNF-α is a pleiotropic cytokine and exerts its biological functions by interactions with two members of the TNF receptor (TNFR)59, both of which trigger several signal transduction pathways, including apoptosis mediated by CASPASE family60,61, the activation of NF-κB and JNK mediated by scaffolding protein TRAF (TNF receptor associated factors)62. Apoptosis, also known as programmed cell death (PCD), is the process of the autonomic death of pathological cell strictly followed by procedure in multicellular organisms, and is regulated by several genes. Among these pathways, the apoptotic genes such as casp3, casp8, tp53i3 and cell cycle inhibiting gene gadd45a were differentially expressed after the knockdown of Cf-SOX2. Combining the TUNEL results in our research, we suggest that the DEGs of apoptotic related pathways may be regulated by Cf-SOX2, which together participate in germ cell apoptosis.
By means of transcriptome analysis, we suggested that the reduction of germ cells in testes after the knockdown of Cf-SOX2 could be resulted from the suppression of cell proliferation, the abnormal expression of spermatogenesis genes and the activation of apoptosis pathways. Several potential downstream genes of Cf-SOX2 were proposed and the subsequent expression characteristics and functional analysis of these genes could be studied in future for revealing detailed regulation mechanisms. Previous study of KLF4 gene in C. farreri indicated that Cf-KLF4 regulates early spermatogenesis, and promotes the differentiation of spermatogonia and spermatocyte, as well as the apoptosis of spermatocytes23. And our further transcriptomic study after Cf-KLF4 was knockdown showed that several genes exhibited similar expression patterns (IAPs, TNFR, TRAF etc.) (unpublished data) as in this study, implying there may exist some correlation between Cf-SOX2 and Cf-KLF4 in testis development. Further studies are needed to explore the interrelation between these two genes, as well as the regulatory mechanism of spermatogenesis.
In the present work, Cf-SOX2 mRNAs were located in spermatogonia, spermatocytes, spermatids and spermatozoa. After knockdown of Cf-SOX2 mRNA in testes, the number of germ cells greatly reduced and a lot of spermatocytes went into apoptosis status, which lead to spermatogenic failure and indicated that Cf-SOX2 played an important role in the development of testis and spermatogenesis. 2,067 DEGs were obtained through transcriptome analysis after the knockdown of Cf-SOX2, and among those DEGs, genes related to the spermatogenesis (ZMYND10, NIPBL, MFGE8, ZFAND3, ARMC3, HTT, TUSC3) were down-regulated, while other genes (CASP2, CASP3, CASP8, BIRC3, SAMD9, CREBZF, IQSEC1, BMP7) involved in cell apoptosis or cell proliferation were up- or down-regulated, indicating that Cf-SOX2 regulates the development of spermatogenic cells and maintains the testis function through multiple biological pathways. Our study provides clues for exploring the regulatory mechanism of SOX2 gene in bivalve testis development.
Materials and methods
Ethics statement
This study was approved by the Animal Ethics Committee of Ocean University of China, Qingdao, China.
Experimental animals
Healthy male and female scallops C. farreri with mean shell height 6.19 ± 0.35 cm were collected from Shazikou Bay (Qingdao, China). The testes were excised, weighted and dissected, part of them were firstly fixed in 4% paraformaldehyde (dissolved in with 0.01 M phosphate buffered saline, PBS) at 4 °C for 24 h, then dehydrated with serial methanol (25%, 50%, 75%, 100%) diluted with 0.01 M PBS and stored in 100% methanol at −20 °C for histology and in situ hybridization. The remainders were immediately frozen in liquid nitrogen and stored at −80 °C for total RNA extraction.
According to histological characteristics and the gonadosomatic index (GSI = gonad weight/soft tissue weight × 100) described by Liao et al.63 and Liu et al.64, the testes of C. farreri used in our study were grouped into three stages, the proliferative stage (GSI = 4.07 ± 0.72), the growing stage (GSI = 6.90 ± 0.58) and the mature stage (GSI = 9.76 ± 1.46).
Histology
Samples were immersed into 100% ethanol twice, cleared in xylene and embedded in paraffin wax. The 5-μm-thick sections were produced according to the procedure described by Liu et al.64. The sections were observed and photographed using a Nikon E80i microscope (Nikon, Tokyo, Japan).
RNA extraction
Total RNAs were extracted using the conventional thiocyanate-phenol-chloroform method65, and were then incubated with DNase I (Takara, Dalian, China) to remove genomic DNA contamination, and purified using the RNeasy mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instruction. The RNA quality and quantity were assessed with electrophoresis and spectrophotometry.
Quantitative RT-PCR (qRT-PCR)
qRT-PCR analysis was performed to determine the expression level of Cf-SOX2 mRNA in testes at different stages. Based on the full-length sequence of Cf-SOX2 (GenBank Acc. No. KF836755.1), two specific primers (Table 1) amplifying a 135 bp product were designed. The C. farreri elongation factor 1 alpha (ef-1α) (GenBank Acc. No. JQ278034.1) was selected as a reference gene66. The amplification was carried out in a 20 μl volume using Roche LightCycler 480 Real-Time PCR System (Roche, Basel, Switzerland) with SYBR Green Master Mix (Takara, Dalian, China) following the manufacturer’s instruction. Data were analyzed using the Roche LightCycler 480 system software version 1.5 (Roche, Basel, Switzerland), and the 2−ΔΔCt method was used to analyze the relative levels of SOX2 mRNA. All data were presented as mean ± SEM from five samples with three parallel repetitions. Besides, all qRT-PCR assays were validated in compliance with “the MIQE guidelines”67.
In situ hybridization (ISH)
The 550 bp DIG-labeled RNA sense and antisense probes of Cf-SOX2 were generated according to Liang et al.29. The 827 bp Cf-VASA probes were generated with the C. farreri VASA gene cDNA sequence (GenBank acc. no. DQ452383) and two specific primers VASA-F: 5′-TAATACGACTCACTATAGGGAGGAAGGACACGAGAACACGCAG-3′ (T7 promoter underlined) and VASA-R: 5′- ATTTAGGTGACACTATAGAAGCGCAGTAATACACGAGGACAGTC-3′ (SP6 promoter underlined). The testes were dehydrated in ethanol, cleared in xylene, embedded in paraffin, and then cut into 5-μm-thick sections for the testes at proliferative stage and the growing stage, and 3-μm-thick sections for mature stage. Sections were fixed to a slide with 0.1% polylysine at 37 °C for 12 h. In situ hybridization was performed as described in Feng et al.68, with modifications of samples digested for 10 min at 37 °C with 2 μg/ml protease K and counter stained with 1% neutral red. Both the Cf-SOX2 and Cf-VASA mRNA were detected for the testes of 3rd day scallops after RNAi, while other samples were detected with Cf-SOX2 probe only. Positive and negative detections were performed using the antisense and sense probe, respectively. The sections were observed and photographed using a Nikon E80i microscope (Nikon, Tokyo, Japan).
RNAi assay
dsRNA synthesis
The procedure was performed as described by Suzuki et al.69 and Yang et al.23 with some modifications. Two specific primers, RNAi-F: 5′-TAATACGACTCACTATAGGGAGAGGACAAATACGCTCTACCAGG-3′ (T7 promoter underlined) and RNAi-R: 5′-TAATACGACTCACTATAGGGAGCATATGAGTAAGCGGAACAG-3′ (T7 promoter underlined) were designed according to Cf-SOX2 cDNA sequence to amplify a fragment of 506 bp. The two primers of EGFP-dsRNA were used as described by Hou et al.70 to amplify a fragment of 497 bp used as control. Gel-purified PCR products were transcribed in vitro using the T7 MEGAscript RNAi Kit (Ambion, Austin, USA) to synthesis double-stranded RNA (dsRNA). The phenol/chloroform-extracted and ethanol-precipitated dsRNAs were suspended in RNase-free 0.01 M PBS (pH 7.4). The integrity and quantity of the dsRNA was assayed with electrophoresis and spectrophotometry.
dsRNA administration and sampling
Healthy male scallops C. farreri with mean shell height 6.28 ± 0.28 cm were purchased from the Nanshan Aquatic Product Market (Qingdao, China). Seventy-five male scallops at the proliferative stage were evenly assigned and raised in three aquaria with 540 L filtered, aerated seawater at 16.0 ± 0.2 °C, respectively. The scallops were fed with unicellular algae Isochrysis galbana and Chaetoceros muelleri and water was renewed daily during the experiment. Eighty microliter PBS containing 30 μg SOX2-dsRNA (SOX2-dsRNA group) or 80 μl PBS containing 30 μg EGFP-dsRNA (EGFP-dsRNA group) were injected into the adductor muscle of male scallops at T0 (initiation of this assay) and T7 (the 7th day), respectively. Both the EGFP-dsRNA and the blank (no injection) groups were used as controls. The testes were weighted and sampled at 3rd day (five scallops for each group), 10th day (three scallops for each group) and 21st day (six scallops for each group) post RNAi, respectively.
Detection of histological feature after RNAi
Testis sections of the sampled scallops from the blank group, SOX2-dsRNA group and EGFP-dsRNA group were made following the method in Section “Histology”. The tissue characteristics of testes and the germ cell types were observed with Nikon E80i microscope (Nikon, Tokyo, Japan).
Quantification and composition of germ cells in the follicles of testes
Testis sections of the scallops from 3rd day were inspected under microscope to determine the quantification and composition of germ cells. All germ cells in the follicles of testes were distinguished according to Liu et al.64 and counted by randomly observing three sights (6400 μm2 for each) in histological sections of testes, and percentage of germ cells at different groups was calculated.
TdT-mediated dUTP Nick-End Labeling (TUNEL) assay
By using a DeadEndTM Colorimetric TUNEL System Kit (Promega, Madison, USA), the TUNEL assay was conducted to detect cell apoptosis in the testes of 3rd day scallops according to the manufacturer’s instruction. Sections of the testis were prepared following the method in Section “Histology”. Observation and digital images were taken with Nikon E80i microscope (Nikon, Tokyo, Japan).
Transcriptome analysis
Sample preparation and library construction for transcriptome sequencing
The testes of three scallops on the 3rd day post RNAi and from each of SOX2-dsRNA group and blank group were used for transcriptome sequencing. The method of total RNA extraction was the same as Section “RNA extraction”.
A total amount of 1.5 µg RNA per sample was used for library preparation. Sequencing libraries were generated using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, Ipswich, USA) following manufacturer’s recommendations. The libraries were subjected to an Illumina Hiseq. 2000 platform with pair-end sequencing of 150 bp.
De novo transcriptome assembly and functional annotation
Raw reads were first preprocessed for quality control to obtain the clean reads by removing reads containing adapters, ambiguous ‘N’ nucleotides (with the ratio of ‘N’ to be more than 10%), and low quality reads (with the Phred quality scores ≤ 20). Then all clean reads from the six libraries were jointly assembled using Trinity software as described for de novo transcriptome assembly71. Unigenes were aligned with diamond and NCBI blast 2.2.28+ to the databases NR, NT and Swiss-Prot with an E-value threshold of 1.0E-5, as well as KOG with an E-value threshold of 1.0E-3 to predict the function of genes. Blast2GO72 software was used to obtain GO annotations. Pathway assignments were determined based on the KEGG database using KAAS (KEGG Automatic Annotation Server, http://www.genome.jp/kegg/kaas/) with an E-value threshold of 1.0E-6.
Differential expression analysis
Gene expression levels for each sample were estimated using RSEM73 and each unigene was normalized by fragments per kilobase of exon model per million mapped reads (FPKM)74. Differential expression analysis of these two groups was performed using the DESeq R package (1.10.1)75. The resulting P values were adjusted using the Benjamini-Hochberg’s false discovery rate (FRD)76. Genes with an FDR < 0.05 found by DESeq were assigned as differentially expressed.
GO and KEGG enrichment analysis
Gene Ontology (GO) enrichment analysis of the differentially expressed genes (DEGs) was implemented by the GOseq R packages based Wallenius non-central hyper-geometric distribution77. KOBAS78 software was utilized to test the statistical enrichment of differential expression genes in KEGG pathways.
qRT-PCR validation
Expression levels of 8 selected DEGs, casp2, casp3, casp8, crebzf, htt, tusc3, bmp7, ddx6 were validated using qRT-PCR with the 6 RNA samples of testes used for the transcriptional analysis. Gene-specific primers (Table 1) were designed using Primer 3 (http://primer3.ut.ee/) based on their ORF sequences. All procedures were identical as in Section “Quantitative RT-PCR (qRT-PCR)”.
Statistical analysis
All data were presented as means ± SEM. Significant differences between means were tested using one-way analysis of variance (ANOVA) followed by Tukey’s HSD test (SPSS software version 18.0; SPSS, Chicago, USA), and the significant level was set at P < 0.05.
Availability of Dupporting Data
Raw sequence reads of RNA-seq can be found in the NCBI Sequence Read Archive (SRP125788).
References
Avilion, A. A. et al. Multipotent cell lineages in early mouse development depend on Sox2 function. Gene Dev. 17, 126–140 (2003).
Keramari, M. et al. Sox2 is essential for formation of trophectoderm in the preimplantation embryo. PloS One 5, e13952 (2010).
Ellis, P. et al. Sox2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev. Neurosci. 26, 148–165 (2005).
Masui, S. et al. Pluripotency governed by SOX2 via regulation of OCT3/4 expression in mouse embryonic stem cells. Nat. Cell Biol. 9, 625–635 (2007).
Liu, K. et al. The multiple roles for SOX2 in stem cell maintenance and tumorigenesis. Cell Signal 25, 1264–1271 (2013).
Ferri, A. L. et al. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 131, 3805–3819 (2004).
Archer, T. C., Jin, J. & Casey, E. S. Interaction of SOX1, SOX2, SOX3 and OCT4 during primary neurogenesis. Dev. Biol. 350, 429–440 (2011).
Cimadamore, F. et al. SOX2-LIN28/let-7 pathway regulates proliferation and neurogenesis in neural precursors. PNAS 110, E3017–E3026 (2013).
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Huangfu, D. et al. Induction of pluripotent stem cells from primary human fibroblasts with only OCT4 and SOX2. Nat. Biotechnol. 26, 1269–1275 (2008).
Montserrat, N. et al. Generation of induced pluripotent stem cells from human renal proximal tubular cells with only two transcription factors, OCT4 and SOX2. J. Biol. Chem. 287, 24131–24138 (2012).
Campolo, F. et al. Essential role of Sox2 for the establishment and maintenance of the germ cell line. Stem Cells 31, 1408–1421 (2013).
Zeng, L. et al. Expression of the Sox2 gene in different tissues and different months on testis of Hyriopsis schlegelii. Acta Hydrobiologica Sinica 36, 205–211 (2012).
Wu, D. et al. Structural characteristics of gonadal development and hermaphroditic phenomenon in freshwater pearl mussel, Sinohyriopisis schlegelii (Hyriopisis schlegelii). Tissue Cell 49, 440–446 (2017).
Gao, J. et al. Identification and characterization of a Sox2 homolog in the Japanese flounder Paralichthys olivaceus. Gene 544, 165–176 (2014).
Patra, S. K. et al. First evidence of molecular characterization of rohu carp Sox2 gene being expressed in proliferating spermatogonial cells. Theriogenology 84, 268–276 (2015).
Phillips, B. T., Gassei, K. & Orwig, K. E. Spermatogonial stem cell regulation and spermatogenesis. Philos. T. R. Soc B. 365, 1663–1678 (2010).
Neuhaus., N. et al. Single-cell gene expression analysis reveals diversity among human spermatogonia. Mol. Hum. Reprod. 23, 79–90 (2017).
Llera-Herrera., R. et al. Identification of make gametogenesis expressed genes from the scallop Nodipecten subnodosus by suppressive subtraction hybridization and pyrosequencing. PloS One 8, e73176 (2013).
Li., H. et al. Characterization, expression and function analysis of DAX1 Gene of scallop (Chlamys farreri Jones and Preston 1904) during its gametogenesis. J. Ocean U. China 13, 696–704 (2014).
Li. H. et al. Expression characteristics of β-catenin in scallop Chlamys farreri gonads and its role as a potential upstream gene of Dax1 through canonical Wnt signaling pathway regulating the spermatogenesis. PloS One 9, e115917.
Liu., X. et al. Gonadogenesis in scallop Chlamys farreri and Cf‐foxl2 expression pattern during gonadal sex differentiation. Aquac. res. 47, 1605–1611 (2016).
Yang, D. et al. A novel role of Krüppel-like factor 4 in Zhikong scallop Chlamys farreri during spermatogenesis. PloS One 12, e0180351 (2017).
Ma., X. et al. Piwi1 is essential for gametogenesis in mollusk Chlamys farreri. PeerJ 5, e3412 (2017).
Zhang, G. F. et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49–54 (2012).
Teaniniuraitemoana, V. et al. Gonad transcriptome analysis of pearl oyster Pinctada margaritifera: identification of potential sex differentiation and sex determining genes. BMC Genomics 15, 491 (2014).
Li, Y. et al. Transcriptome Sequencing and Comparative Analysis of Ovary and Testis Identifies Potential Key Sex-Related Genes and Pathways in Scallop Patinopecten yessoensis. Mar. Biotechnol. 18, 453–465 (2016).
He, Y. et al. Molecular cloning and characterization of SoxB2 gene from Zhikong scallop Chlamys farreri. Chin. J. Oceanol. Limn. 31, 1216–1225 (2013).
Liang, S., Ma, X., Han, T., Yang, D. & Zhang, Z. Expression pattern of Chlamys farreri Sox2 in eggs, embryos and larvae of various stages. J. Ocean U. China 14, 731–738 (2015).
Yu, F., Wang, M., Gui, J., Zhou, L. & Yu, X. Molecular cloning and expression patterns of Sox11 gene in Pinctada martensii. Acta Hydrobiologica Sinica 40, 71–75 (2016).
Li, C. F. et al. Human sterile alpha motif domain 9, a novel gene identified as down-regulated in aggressive fibromatosis, is absent in the mouse. BMC Genomics 8, 92 (2007).
Bergeron, T. et al. The effect of ZHANGFEI on the unfolded protein response and growth of cells derived from canine and human osteosarcomas. Vet. Comp. Oncol. 11, 140–150 (2013).
Morcillo, J., Martínez-Morales, J. R., Trousse, F., Fermin, Y. & Sowden, J. C. Proper patterning of the optic fissure requires the sequential activity of BMP7 and SHH. Development 133, 3179–3190 (2006).
Someya, A., Moss, J. & Nagaoka, I. Involvement of a guanine nucleotide-exchange protein, ARF-GEP100/BRAG2a, in the apoptotic cell death of monocytic phagocytes. J. Leukocyte Biol. 80, 915–921 (2006).
Valderrama, X., Rapin, N., Verge, V. M. K. & Misra, V. Zhangfei induces the expression of the nerve growth factor receptor, TRKA, in medulloblastoma cells and causes their differentiation or apoptosis. J. Neuro-Oncol. 91, 7–17 (2009).
Guo, Y., Srinivasula, S. M., Druilhe, A., Fernandes-Alnemri, T. & Alnemri, E. S. Caspase2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J. Biol. Chem. 277, 13430–13437 (2002).
Hoshi, T. et al. Immunohistochemistry of Caspase3/CPP32 in human stomach and its correlation with cell proliferation and apoptosis. Anticancer Res. 18, 4347–4353 (1998).
Kuwana, T. et al. Apoptosis induction by Caspase8 is amplified through the mitochondrial release of cytochrome c. J. Biol. Chem. 273, 16589–16594 (1998).
Sanchez, I. et al. Caspase8 is required for cell death induced by expanded polyglutamine repeats. Neuron 22, 623–633 (1999).
Susin, S. A. et al. Mitochondrial release of caspase2 and 9 during the apoptotic process. J. Exp. Med. 189, 381–394 (1999).
Wang, Y., Zhou, Q., Wang, S. & Zhang, J. Homocysteine-induced apoptosis of endothelial cell and its antagonism by folic acid–the roles of caspase3, c-IAP1 and c-IAP2. J. Hygiene Res. 33, 310–313 (2004).
Deveraux, Q. L. & Reed, J. C. IAP family proteins-suppressors of apoptosis. Gene Dev. 13, 239–252 (1999).
Salvesen, G. S. & Duckett, C. S. IAP proteins: blocking the road to death’s door. Nat. Rev. Mol. Cell Bio. 3, 401–410 (2002).
Tamm, I. et al. IAP-family protein survivin inhibits caspase activity and apoptosis induced by FAS (CD95), BAX, caspases, and anticancer drugs. Cancer Res. 58, 5315–5320 (1998).
Agathanggelou, A. et al. Epigenetic inactivation of the candidate 3p21. 3 suppressor gene Blu in human cancers. Oncogene 22, 1580 (2003).
McClintock, T. S. & Glasser, C. E. Bos,e S.C. & Bergman, D.A. Tissue expression patterns identify mouse cilia genes. Physiol. Genomics 32, 198–206 (2008).
Moore, D. J. et al. Mutations in Zmynd10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 93, 346–356 (2013).
Pausch, H. A frameshift mutation in ARMC3 is associated with a tail stump sperm defect in Swedish Red (Bos taurus) cattle. BMC Genetics 17, 49 (2016).
Kuleszewicz, K., Fu, X. & Kudo, N. R. Cohesin loading factor Nipbl localizes to chromosome axes during mammalian meiotic prophase. Cell Div. 8, 12 (2013).
Copland, S. D., Murphy, A. A. & Shur, B. D. The mouse gamete adhesin, SED1, is expressed on the surface of acrosome-intact human sperm. Fertil. Steril. 92, 2014–2019 (2009).
Ensslin, M. A. & Shur, B. D. Identification of mouse sperm SED1, a bimotif EGF repeat and discoidin-domain protein involved in sperm-egg binding. Cell 114, 405–417 (2003).
de Luis, O., López-Fernández, L. A. & del Mazo, J. Tex27, a gene containing a zinc-finger domain, is up-regulated during the haploid stages of spermatogenesis. Exp. Cell Res. 249, 320–326 (1999).
Ma, K., Liao, M., Liu, F., Ye, B. & Sun, F. Charactering the Zfand3 gene mapped in the sex-determining locus in hybrid tilapia (Oreochromis spp.). Sci. Rep. 6, 25471 (2016).
Otake, S., Endo, D. & Park, M. K. Molecular characterization of two isoforms of Zfand3 cDNA from the Japanese quail and the leopard gecko, and different expression patterns between testis and ovary. Gene 488, 23–34 (2011).
Khalid, A. M., Asano, A., Hosaka, Y. Z., Takeuchi, T. & Yamano, Y. Tumor suppressor candidate TUSC3 expression during rat testis maturation. Biosci. Biotech. Bioch. 77, 2019–2024 (2013).
Dragatsis, I., Levine, M. S. & Zeitlin, S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 26, 300–306 (2000).
Leavitt, B. R. et al. Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am. J. Hum. Genet. 68, 313–324 (2001).
Vousden, K. H. & Lu, X. Live or let die: the cell’s response to p53. Nat. Rev. Cancer 2, 594 (2002).
Yan, Z. et al. A mutated human tumor necrosis factor-alpha improves the therapeutic index in vitro and in vivo. Cytotherapy 8, 415–423 (2006).
Harper, N., Hughes, M., MacFarlane, M. & Cohen, G. M. Fas-associated death domain protein and caspase-8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during tumor necrosis factor-induced apoptosis. J. Biol. Chem. 278, 25534–25541 (2003).
Micheau, O. & Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003).
Till, A. et al. The Met-196→Arg variation of human tumor necrosis factor receptor 2 (TNFR2) affects TNF-α-induced apoptosis by impaired NF-κB signaling and target gene expression. J. Biol. Chem. 280, 5994–6004 (2005).
Liao, C., Xu, Y. & Wang, Y. Reproductive cycle of the scallop Chlamys farreri (Jones and Preston) at Qingdao. J. Fish. China. 7, 1–13 (1983).
Liu, J. et al. Variations of estradiol-17β and testosterone levels correlated with gametogenesis in the gonad of Zhikong scallop (Chlamys farreri) during annual reproductive cycle. Can. J. Zool. 92, 195–204 (2014).
Chomczynski, P. & Sacchi, N. Single-step method of RNA isolation by acid guanidinium thocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159 (1987).
Zhou, Z. et al. The expression of dopa decarboxylase and dopamine beta hydroxylase and their responding to bacterial challenge during the ontogenesis of scallop Chlamys farreri. Fish Shellfish Immun. 33, 67–74 (2012).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 (2009).
Feng, Z. F., Zhang, Z. F., Shao, M. Y. & Zhu, W. Developmental expression pattern of the Fc-vasa-like gene, gonadogenesis and development of germ cell in Chinese shrimp, Fenneropenaeus chinensis. Aquaculture 314, 202–209 (2011).
Suzuki, M. et al. An acidic matrix protein, PIF, is a key macromolecule for nacre formation. Science 325, 1388–1390 (2009).
Hou, F. et al. RNAi knock-down of shrimp Litopenaeus vannamei Toll gene and immune deficiency gene reveals their difference in regulating antimicrobial peptides transcription. Dev. Comp. Immunol. 44, 255–260 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Trapnell, C. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist Soc. B 57, 289–300 (1995).
Young, M. D., Wakefield, M. J., Smyth, G. K. & Oshlack, A. Gene ontology analysis for RNA-Seq: accounting for selection bias. Genome Biol. 11, R14 (2010).
Mao, X., Cai, T., Olyarchuk, J. G. & Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793 (2005).
Acknowledgements
This work was support by the Shandong Provincial Natural Science Foundation, China (ZR2016CQ24), and the National High Technology Research and Development Program of China (2012AA10A402).
Author information
Authors and Affiliations
Contributions
S.L., Z.Z. and Z.Q. conceived and designed the experiments; S.L., D.L., X.L. and X.Y. performed the experiments; S.L., M.W., and Q.L. analyzed the data; D.L., M.W., X.Y. and H.M. contributed materials/analysis tools; S.L. and Z.Z. wrote the paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liang, S., Liu, D., Li, X. et al. SOX2 participates in spermatogenesis of Zhikong scallop Chlamys farreri. Sci Rep 9, 76 (2019). https://doi.org/10.1038/s41598-018-35983-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-35983-3
This article is cited by
-
A signal recognition particle receptor gene from the sea cucumber, Apostichopus japonicas
Scientific Reports (2023)
-
Proteome and Transcriptome Analysis of Gonads Reveals Intersex in Gigantidas haimaensis
BMC Genomics (2022)
-
Dynamic transcriptome analysis reveals the gene network of gonadal development from the early history life stages in dwarf surfclam Mulinia lateralis
Biology of Sex Differences (2022)
-
Genetic targeting or pharmacological inhibition of galectin-3 dampens microglia reactivity and delays retinal degeneration
Journal of Neuroinflammation (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
