
Abstract
Glaulactams A–C (1–3), which possess a novel skeleton, as well as the known compound daphmanidin B (4), were isolated from the leaves of Daphniphyllum glaucescens and separated using ion-exchange chromatography aided by NMR fingerprinting. Their structures, including their absolute configurations, were elucidated by spectroscopic analyses and time-dependent density-functional-theory-calculated electronic circular dichroism spectra; the data were subsequently analyzed to gain insight into the respective biogenetic relationships between the isolates, which exhibited anti-H1N1 and immunosuppressive activities.
Similar content being viewed by others


Introduction
The first daphniphylline-type alkaloid, daphniphylline, which possesses a polycyclic, C30-aliphatic structure, was discovered in 19661. A number of novel structures from the Daphniphyllaceae family of plants were recently reported, including himalensine A, with its 13,14,22-trinorcalyciphylline A backbone2, himalensine B with its 22-nor-1,13-secodaphnicyclidin framework2, and macropodumines A–C with fused pentacyclic ring systems3. The complex and fascinating structures of these alkaloids render them synthetically challenging4. Many types of alkaloid are reported to be produced by Daphniphyllum glaucescens, including daphniglaucin C, which has a novel structure that contains hexahydroazulene and octahydroindole ring systems5 and daphniglaucins A and B, which have unique 1-azoniatetracyclo[5.2.2.01,60.4,9]undecane motifs6. In addition, daphniglaucins D–H possess fused hexacyclic skeletons, and daphniglaucins J and K are yuzurimine-type alkaloids that have previously been reported from this species7.
Natural product chemists are very interested in the discovery of novel structures with unique properties8. To date, a variety of methods have been used to facilitate the discovery of novel natural products, with examples including NMR-fingerprinting8 and LC-MS-guided methods9. Daphnezomine-F-type10,11 and daphmanidin-C-type12 alkaloids are rare groups of daphniphyllum alkaloids; they each possess a lactam functional group that is biogenetically derived through the oxidative cleavage of a yuzurimine C–C bond. The presence of lactams in these daphniphyllum compounds inspired us to develop a new method that combines ion-exchange chromatography (IXC) and NMR fingerprinting to screen for new alkaloids. Hence, in this article, we report the isolation, structural characterization, and biological evaluation of glaulactams A–C (1–3) from the leaves of Daphniphyllum glaucescens.
Results and Discussion
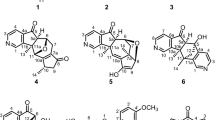
Air-dried D. glaucescens leaves (1.53 kg) were extracted with MeOH and concentrated to obtain a crude extract, which was dispersed in 80% aqueous MeOH to give a methanolic suspension. This suspension was then partitioned with hexane (4 × 2 L) to remove the lipid constituents. The methanolic extract was filtered and passed through a cation-exchange resin. The eluent containing the acidic and neutral compounds was concentrated under reduced pressure and subjected to Diaion HP20 column chromatography, followed by vacuum liquid chromatography (VLC) over silica gel to yield 26 fractions (4A–4Z); among these fractions, the 1H NMR spectra of fractions 4V, 4W, and 4Z were found to exhibit signals characteristic of daphniphyllum alkaloids (see the Extraction and Isolation section). As a result, these three fractions were selected for repeated column chromatography over silica gel and RP-18 stationary phases to yield three compounds that had structures that were consistent with 1 (4.2 mg), 2 (29.2 mg), and 3 (3.0 mg) (Fig. 1), as described below.

Structures of compounds 1–3.
Glaulactam A (1) exhibited a molecular ion peak at m/z 486.2494 [M + H]+ (calcd., 486.2486), consistent with the C27H35NO7 molecular formula and 11 degrees of unsaturation. The IR spectrum of 1 exhibited bands corresponding to carbonyl functionalities (1736, 1730, 1712, and 1695 cm−1). The 13C and 1H NMR data (Table 1) reveal the presence of 27 carbon signals ascribed to two acetyl groups (δC 168.9, 20.1, δH 2.05; δC 170.5, 20.4, δH 2.07), a methyl carboxylate (δC 173.9, 50.4, δH 3.59), a conjugated diene (δC 120.9, 134.1, 134.6, 143.3, δH 5.35), an acetoxy-bearing methine (δC 76.6, δH 5.51), an acetoxy-bearing methylene (δC 66.4, δH 4.06, 5.03), and an amide carbonyl (δC 180.5). The above functionalities accounted for six out of the 11 degrees of unsaturation, suggesting that 1 is pentacyclic. The HSQC, 1H–1H COSY, and HSQC-COSY spectra of 1 reveal three structural connectivities, namely I (H-2 to H-18, H-2 to H-4, and H-18 to both H2-19 and H3-20), II (H-6 to H2-7, H-11 to H2-12, and H-6 to H2-12), and III (H2-13 to H2-17), as is illustrated in Fig. 2.

Selected 1H–1H COSY and HMBC correlations in 1–3.
Subsequent HMBC spectral analysis revealed where these three structural fragments are linked. The C-1 carbonyl (δC 180.5) and C-19 (δC 51.4, δH 3.16, m, 2 H) of fragment I were speculated to be due to a γ-lactam ring, which was verified by the observed HMBC-derived H2-3/C-1 and H2-19/C-1 correlations. HMBC correlations between H2-21 and C-4, C-5, C-6, and C-8, and between H2-7 and C-1 and C-19 linked fragment I to II, resulting in an amide-bridged bicyclic system (C-1 to C-7 and the N atom, and C-18 to C-19). The link between the methyl carboxylate and C-14 in fragment III was made on the basis of the HMBC correlations between both H-14 and H3-23, and C-22, while C-13, C-15, and C-17 are attached to C-8, C-9, and C-10 of the conjugated diene on the basis of the HMBC correlations between H-14 and C-8 and C-9, and between H-11 and C-9 and C-17. The acetoxy groups at C-4 and C-21 were assigned on the basis of HMBC correlations between H-4 and H2-21 and the respective acetyl carbonyls at δC 168.9 and 170.5. Hence, the planar structure of 1 was assigned to be a fused pentacyclic ring with a γ-lactam functionality (Fig. 2).
The relative configuration of 1 was deduced through NOE-correlation analysis, as indicated in the Chem3D drawing (Fig. 3); H-4/H-19, H-4/H-6, H-4/H3-20, H-21a/H-12b, and H-6/H2-12 NOE correlations indicate that H2-21, H-6, H3-20, and H-4 are cofacial and were arbitrarily assigned to be β-oriented. In addition, the NOE spectrum reveals a correlation between H-2 and H2-3, in addition to a correlation between H-3a and H3-20, implying that H-2 is positioned between H2-3, but opposite to H3-20 (Fig. 3). In addition, H-13a/H-21b, H-13b/H-14, and H-14/H-15 correlations suggest that both H-14 and H-15 are α-oriented.

Selected NOESY correlations in 1 and 3.
High-resolution electrospray-ionization mass spectrometry (HRESIMS) revealed that glaulactam B (2) has the molecular formula C25H33NO5, the mass of which is less than that of 1 by one CH2CO2 unit. In addition, the 13C and 1H NMR data for 2 are similar to those of 1; however, instead of possessing an acetoxy-bearing methine, as in 1, an sp3 methylene is present at C-4 (δC 36.3) in 2 (Table 1). This was confirmed by the HMBC correlations between H2-21 and C-4, C-5, C-6, and C-8 (Fig. 2). The planar structure of 2 (Fig. 2) was established following 1H-1H COSY, HSQC-TOCSY, and HMBC spectral analyses. With the exception of C-4, the same relative configurations were assigned to 1 and 2 on the basis of detailed NOE-correlation analyses (Supplementary Fig. S4).
Glaulactam C (3) was subjected to HRESIMS, which suggested that its molecular formula was C25H31NO5, which contains two less hydrogen atoms than 2. IR absorptions at 1735, 1701, 1687, and 1641 cm−1 indicate the presence of carbonyl and alkene functionalities. The 1H and 13C NMR spectra of 3 reveal the presence of characteristic signals that are attributable to a similar γ-lactam ring (δC 181.7, 45.5, 27.9, 51.6, and 12.4), a 21-acetoxy moiety (δC 77.0, 170.2, and 20.3), and a 14-methyl carboxylate (δC 164.9, 50.3) functionality as was observed for 2. Moreover, the presence of six olefinic-carbon signals (δC 119.4, 126.7, 134.2, 148.7, 160.2, and 168.2; δH 7.03) and an upfield-shifted carboxylate carbon (δC 164.9) that is correlated to the methyl protons (δH 3.75) in the HMBC spectrum, suggest that the carboxylate is conjugated to the three alkene units (Fig. 2). The HMBC correlations between H-13 and C-8, C-9, C-14, and C-15, and between H2-16 and C-9 and C-10, as well as H2-11 and C-9, C-10, and C-17, led to the assignment of a fulvene moiety, as illustrated in Fig. 2. The planar structure of 3 was confirmed by further HSQC-TOCSY spin-system analyses (H-6 to H2-7, H-11 to H2-12, and H-6 to H2-12) and HMBC correlations between H2-21 and C-4, C-5, C-6, and C-8. The H-21b/H-6, H-19b/H3-20, and H-19b/H-6 NOE correlations imply that H-6, H2-21, and H3-20 are oriented on the same face of the molecule. Moreover, H3-20/H-3a, H-2/H-3a, and H-2/H-3b correlations suggest that H-2 is positioned opposite to H3-20 (Fig. 3).
The absolute configurations of compounds 1–3 were determined by comparing the experimental electronic circular dichroism (ECD) spectra to those calculated theoretically. Compounds 1–3 were subjected to standard conformational analysis as implemented in the Confab program13. The generated lowest-energy structures following further B3LYP/6–31G(d) optimizations were then used to calculate ECD spectra by time-dependent density functional theory (TDDFT) at the TD-CAM-B3LYP/def2TZVP level. All calculations were performed using the Gaussian 09 Rev. D program package using the “ultrafine grid” option, Integral (Grid = UltraFine), with solvent effects accounted for using the IEFPCM method14. The ECD spectrum of each compound 1–3 was finally generated as the Boltzmann-weighted sum of the spectra generated for the various conformers in each case, which resulted in the establishment of the absolute configurations of glaulactams A–C were, as illustrated by structures 1–3, since the calculated spectra are in good agreement with those acquired experimentally (Fig. 4; Supplementary Fig. S5).

Experimental (red) and calculated (blue) ECD spectra of 1.
It is possible to describe the biosynthetic origins of 1–4 on the basis of the lactam formation mechanism of hemiaminals, the structure of yuzurimine E15 and a proposed compound i. In the proposed mechanism (Fig. 5), oxidative cleavage of the C-1–C-2 bond in yuzurimine E via pathway a16 yields intermediate ii, which, after oxidation at C-2, gives 4. Similarly, C-1–C-8 bond cleavage via pathway b results in the formation of intermediate iii; subsequent deprotonation of the iminol affords lactam 2, which then undergoes oxidation at C-4 and acetylation to yield 1. Double-bond migration and dehydrogenation then result in the transformation of 2 into fulvene 3.

Plausible biogenetic pathways for 1–4.
In preliminary biological screening, the extract of D. glaucescens exhibited antiviral activity against the influenza virus and was immunosuppressive in lipopolysaccharide (LPS)-stimulated murine dendritic cells (DCs). Compounds 1–4 were tested for their anti-influenza virus (i.e., anti-H1N1) activities in Madin-Darby canine kidney (MDCK) cells using the plaque assay with betulinic acid as the positive control17. Cytotoxicity testing revealed that the isolates were not toxic to uninfected host MDCK cells at a concentration of 100 μM (Fig. 6a). However, at a concentration of 50 μM, compounds 1 and 4 were found to substantially inhibit plaque formation of MDCK cells by H1N1 virus infection, to values of 24.4% and 28.0%, respectively. Although less effective under the same treatment conditions, compounds 2 and 3 were found to moderately inhibit plaque formation (69.1% and 63.5%, respectively) (Fig. 6b). In addition, compounds 1–3 were also evaluated for their immunosuppressive activities. Mouse bone-marrow DCs were treated with compounds 1–3, and the immunosuppressive agent quercetin was used as the positive control18. In preliminary studies, compounds 1–3 (50 μg/mL) and quercetin (50 μM) were found to have no significant cytotoxic effects on murine DCs in the presence of LPS (100 ng/mL) (Fig. 7). However, subsequent experiments revealed that compounds 1–3 significantly suppress the levels of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-12p70, and nitric oxide (NO) in LPS-stimulated murine DCs (Fig. 7). These results confirm that the immunosuppressive properties of compounds 1–3 are not due to their cytotoxicities in DCs, and that the observed effects are similar to that induced by quercetin.

Cytotoxic (a) and anti-H1N1(b) activities of compounds 1–4. Betulinic acid was used as the positive control.

(a) Cytotoxicities toward murine bone marrow-derived DCs in the presence of 100 ng/mL LPS for 24 h using the CCK-8 cell-counting assay. (b) TNF-α, (c) IL-6, and (d) IL-12 p70 cytokine levels determined by ELISA, and (e) NO determined using the Griess reagent. Data are expressed as means ± standard deviation (n = 3). Compounds 1–3 were used at 50 μg/mL and quercetin, the positive control, was used at 50 μM.
In summary, IXC and NMR fingerprinting were used to identify and isolate three novel daphniphyllum alkaloids, whose anti-influenza and immunosuppressive activities were then explored. The method described herein can be implemented as a convenient alternative to existing methods commonly employed to extract unique compounds from a complex array of natural products.
Methods
Ethical Statement
The Institutional Animal Care and Use Committee (IACUC) of National Chung Hsing University approved the experimental procedures (approved protocol no. NCHU-IACUC-104-027). The methods were performed in accordance with the approved guidelines.
General experimental procedures
Optical rotations were measured on a JASCO P2000 digital polarimeter and IR spectra were acquired on a Shimadzu IR Prestige-21 FT-IR spectrometer. NMR spectra, in pyridine-d5, were recorded on a 500 MHz Avance III spectrometer (Bruker, Rheinstetten, Germany). The 1H and 13C NMR chemical shifts were referenced to the solvent residual peaks at δH 7.58 and δC 135.5 for pyridine-d5. HRESIMS was performed on an LTQ Orbitrap XL mass spectrometer. Silica gel 60 (Merck, 230−400 mesh), Diaion HP-20 (Supelco™, Bellefonte, PA, USA), and RP-18 gel (LiChroprep 40–63 μm, Merck) were used for column chromatography. A Shimadzu LC-20AT pump and Shimadzu SPD-M20A diode array detector (Shimadzu Inc., Kyoto, Japan) equipped with an Inertsil ODS-3 column (5 µm, 250 × 10 mm, GL Science Inc., Tokyo, Japan) were used for HPLC. Preparative medium-pressure liquid chromatography (MPLC) was performed on an Interchim PuriFlash XS 420 chromatographic system.
Plant Material
The leaves of D. glaucescens subsp. oldhamii were collected in Yilan County, Taiwan in October 2014. The plant was identified by Dr. S.-Y. Hwang. A voucher specimen (specimen no. DG-Chao004) was deposited at the Chinese Medicine Research and Development Center, China Medical University Hospital.
Extraction and Isolation
Air-dried leaves of D. glaucescens (10.38 kg) were exhaustively minced and extracted with MeOH (4 × 20 L). The solvent was concentrated, and 20% water was added to yield an aqueous suspension, to which hexane (4 × 2 L) was added to removed lipids and chlorophylls. The methanolic solution was passed through a strong cation-exchange resin (Dowex 50WX4) to adsorb alkaloids (Supplementary Fig. S6). The collected eluent, which contained nonalkaloid constituents (1530 g), was fractionated by column chromatography (CC; Diaion HP-20: column size: 12.0 × 25.0 cm; successively eluted with 10 L of 30%, 60%, 80%, and 100% MeOH in H2O) to yield four fractions. Fraction 4 (24.95 g), which was eluted with 100% MeOH, was fractionated by silica-gel CC (column size: 6.0 × 15.0 cm; eluted with 9:1 to 0:1 hexane:EtOAc) to yield 26 subfractions (4A–4W). Subfraction 4V (203 mg), which exhibited 1H NMR signals characteristic of daphniphyllum alkaloids (i.e., few methyl signals in the 1–2 ppm region, aliphatic signals at 2–3 ppm, and CH-N signals between 3 and 4 ppm; see Supplementary Fig. S6), were selected for further purification by MPLC (PuriFlash Column, 30 µm, PF-30C18XS/55G; eluted with 70% to 100% MeOH in H2O) to yield 1 (4.2 mg) and 2 (29.2 mg). Subfraction 4 W (288 mg) was fractionated by MPLC (PuriFlash Column, 30 µm, PF-30C18XS/55G; 60% to 100% MeOH in H2O) to give six subfractions (4W1 to 4W6), of which 4W6 was found to possess signals characteristic of daphniphyllum alkaloids. Compound 4 (2.8 mg) was isolated from subfraction 4W6 by HPLC (65% MeOH in H2O). Subfraction 4Z (2.0 g) was chromatographed on silica gel (1:1 to 0:1 hexane:EtOAc), followed by MPLC (PuriFlash Column, 30 µm, PF-30C18XS/55G; 70% to 100% MeOH in H2O) to yield subfraction 4Z2A; the 1H NMR signals of subfraction 4Z2A were found to be characteristic of daphniphyllum alkaloids. Further purification of subfraction 4Z2A by HPLC (65% MeOH in H2O) yielded compound 3 (3.0 mg).
Glaulactam A (1): colorless oil; [α] + 102 (c 0.42, MeOH); ECD (EtOH) λmax (∆ε) 231 (−8.61), 270 (+12.81); UV (MeOH) λmax (log ε) 260 (3.74), 270 (3.81), 280 (3.71) nm; IR (KBr) vmax 2949, 2933, 1736, 1730, 1712, 1695, 1433, 1371, 1247, 1224, 1195, 1168, 1028, and 754 cm−1; 13C and 1H NMR data, see Table 1; ESIMS m/z 486 [M + H]+, 508 [M + Na]+; HRESIMS m/z 486.2494 [M + H]+ (calcd. for C27H36NO7, 486.2486).
Glaulactam B (2): colorless oil; [α] + 146 (c 0.58, MeOH); ECD (EtOH) λmax (∆ε) 229 (−7.33), 270 (+5.40); UV (MeOH) λmax (log ε) 261 (3.67), 271 (3.74), 280 (3.63) nm; IR (KBr) vmax 2949, 2931, 1734, 1699, 1685, 1436, 1375, 1363, 1244, 1193, 1166, 1028, and 752 cm−1; 13C and 1H NMR data, see Table 1; ESIMS m/z 428 [M + H]+, 450 [M + Na]+; HRESIMS m/z 428.2436 [M + H]+ (calcd. for C25H34NO5, 428.2431).
Glaulactam C (3): colorless oil; [α] + 70 (c 0.30, MeOH); ECD (EtOH) λmax (∆ε) 226 (−9.02), 287 (+3.21); UV (MeOH) λmax (log ε) 245 (4.07), 288 (3.91) nm; IR (KBr) vmax 2926, 1735, 1701, 1687, 1641, 1597, 1436, 1365, 1284, 1234, 1195, 1172, 1118, 1062, 1028, and 754 cm−1; 13C and 1H NMR data, see Table 1; ESIMS m/z 448 [M + Na]+; HRESIMS m/z 448.2090 [M + Na]+ (calcd. for C25H31NO5Na, 448.2094).
Cell culture and cytotoxicity assay
Madin-Darby canine kidney (MDCK) cells, (obtained from Dr. Lin, JH, Centers for Disease Control, Taipei, Taiwan) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Hyclone, GE Healthcare Life science) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Hyclone, GE Healthcare Life science) and maintained at 37 °C in a 5% CO2 atmosphere. For the cytotoxicity assay, MDCK cells were seeded in 96-well microplates (1 × 104 cells/well), and cultured for 24 h. Culture media were then replaced with media containing each of the indicated compounds for an additional 72 h. Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-5-(3- carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H tetrazolium (MTS) assay kit purchased from Promega (Madison, WI).
Plaque-reduction assay
Monolayer MDCK cells were seeded in six-well plates (5 × 105 cells/well) for 24 h. The influenza virus A/WSN/33(H1N1) (100 plaque-forming units (PFU) per well) was mixed with each of the indicated compounds for 30 min at room temperature. The mixtures were subsequently adsorbed to the preseeded cells for 1 h at 37 °C. After removal of the medium, the cells were washed with PBS (three times) and then overlaid with 0.3% agarose containing the indicated compounds for an additional 48 h at 37 °C, after which the cells were fixed with 10% formaldehyde for 1 h. Viral plaques were counted by staining with 0.5% crystal violet.
Mice and the preparation of bone marrow-derived murine dendritic cell (DC)
According to the published method19, the murine bone-marrow-derived DCs were prepared from female C57BL/6 mice housed under controlled-temperature (22 ± 2 °C) and humidity (45–65%) conditions, with a 12-h light/dark cycle and free access to food and water. The animals were treated according to the requirements of the Institutional Animal Care and Use Committee of National Chung-Hsing University.
Cell viability assay
DC-cell viability was measured using a cell-counting-kit assay (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan)20, at an absorbance wavelength of 450 nm.
Cytokine and nitric oxide (NO) assay
After centrifugation at 1000 × g for 15 min at 4 °C, the levels of tumor necrosis factor (TNF)-α (cat. no. 900-K54; PeproTech, Inc., London, UK), interleukin (IL)-6 (cat. no. 900-K50; PeproTech, Inc., London, UK), and IL-12p70 (cat. no. 900-K97; PeproTech, Inc., London, UK) in the culture supernatants were determined using murine ELISA kits for the respective cytokine according to the manufacturer’s protocol. The levels of NO were measured indirectly by determining the concentration of NO2- using a spectrophotometric assay based on the Griess reaction.
Computational details
Since different conformers of a specific stereochemical configuration can give different ECD spectra, it is critical to identify all relevant conformers in order to accurately predict the ECD spectrum. Therefore, standard conformational analyses were performed using the Confab program13. All optimized conformations were in a 10 kcal/mol energy window, with a root mean square (RMS) step-size of 0.2 Å. These conformers were then re-optimized at the B3LYP/6-31G(d) level of theory and verified to be true minima on the potential energy surface by frequency analyses. Then resulting geometries were subsequently used for three single-point calculations. CAM-B3LYP/TZVP21 calculations in the vacuum state were carried out in order to obtain converged wavefunctions for the ground states, which were used in the next two calculations. Energies were calculated at the M062x/Def2TZVP level with ethanol as the solvent, as this method provides more precise energies for conformational ordering22. The series of conformers was restricted by removing duplicates (RMSD < 0.2) and conformations outside of a 4 kcal/mol energy window, based on the Gibbs free energy at the M062x/Def2TZVP//B3LYP/6-31G(d) level in ethanol. The resulting structures were used in TDDFT ECD calculations, in ethanol as the solvent, at the TD-CAM-B3LYP/def2TZVP level by considering the 100 lowest-energy states. All calculation were performed using the Gaussian 09 program package with the “ultrafine grid” option, Integral (Grid = UltraFine), and all solvent effects were accounted for using the IEFPCM method14,23. Finally, the overall ECD spectra were combined following Boltzmann weighting on the basis of the Gibbs free energies of the corresponding conformer calculated at the M062x/Def2TZVP//B3LYP/6-31G (d) level. All ECD spectra were processed with SpecDis and simulated by Gaussian functions with bandwidths (σ) of 0.16 eV and by considering velocity representations24.
References
Sakabe, N. et al. Isolation of three new alkaloids from daphniphyllum macropodum Miquel. Tetrahedron Lett. 7, 963–964 (1966).
Zhang, H. et al. Himalensines A and B, alkaloids from Daphniphyllum himalense. Org. Lett. 18, 1202–1205 (2016).
Zhang, W., Guo, Y. W. & Krohn, K. Macropodumines A–C: novel pentacyclic alkaloids with an unusual skeleton or zwitterion moiety from Daphniphyllum macropodum Miq. Chem. Eur. J. 16, 5122–5127 (2006).
Chattopadhyay, A. K. & Hanessian, S. Recent progress in the chemistry of daphniphyllum alkaloids. Chem. Rev. 117, 4104–4146 (2017).
Morita, H. et al. Daphniglaucin C, a novel tetracyclic alkaloid from Daphniphyllum glaucescens. Tetrahedron Lett. 45, 901–904 (2004).
Kobayashi, J. et al. Daphniglaucins A and B, novel polycyclic quaternary alkaloids from Daphniphyllum glaucescen. Org. Lett. 5, 1733–1736 (2003).
Takatsu, H. et al. Daphniglaucins D–H, J, and K, new alkaloids from Daphniphyllum glaucescens. Tetrahedron 60, 6279–5284 (2004).
Grkovic, T. et al. NMR fingerprints of the drug-like natural-product space identify iotrochotazine A: a chemical probe to study Parkinson’s disease. Angew. Chem. Int. Ed. 53, 6070–6074 (2014).
Geng, C. A. et al. LC-MS guided isolation of (±)-sweriledugenin A, a pair of enantiomeric lactones, from Swertia leducii. Org. Lett. 16, 370–373 (2014).
Kobayashi, J. & Kubota, T. The Daphniphyllum alkaloids. Nat. Prod. Rep. 26, 936–962 (2009).
Morita, H., Yoshida, N. & Kobayashi, J. Daphnezomines F and G, novel alkaloids with 1-azabicyclo[5.2.2]undecane moiety from. Daphniphyllum humile. J. Org. Chem. 65, 3558–3562 (2000).
Morita, H. et al. Daphmanidins C and D, novel pentacyclic alkaloids from Daphniphyllum teijsmanii. Org. Lett. 7, 459–462 (2005).
O’Boyle, N. M. et al. Open Babel: an open chemical toolbox. J. Cheminform. 3, 8 (2011).
Cancès, E., Mennucci, B. & Tomasi, J. A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 107, 3032–3041 (1997).
Citar, H. E. et al. New alkaloids from Daphniphyllum calycinum. J. Nat. Prod. 67, 1094–1099 (2004).
Khusnutdinova, J. R., Ben-David, Y. & Milstein, D. Oxidant-free conversion of cyclic amines to lactams and H2 using water as the oxygen atom source. J. Am. Chem. Soc. 136, 2998–3001 (2014).
Hong, E. H. et al. Anti-influenza activity of betulinic acid from Zizyphus jujube on influenza A/RP/8 virus. Biomol. Ther. 23, 345–349 (2015).
Huang, R. Y. et al. Immunosuppressive effect of quercetin on dendritic cell activation and function. J Immunol. 184, 6815–6821 (2010).
Lin, S. et al. Formulated extract from multiple citrus peels impairs dendritic cell functions and attenuates allergic contact hypersensitivity. Int. Immunopharmacol. 20, 12–23 (2014).
Chung, T. W. et al. Sinulariolide suppresses LPS-induced phenotypic and functional maturation of dendritic cells. Mol. Med. Rep. 16, 6992–7000 (2017).
Yanai, T., Tew, D. P. & Handy, N. C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 393, 51–57 (2004).
Walker, M. et al. Performance of M06, M06-2X, and M06-HF density functionals for conformationally flexible anionic clusters: M06 functionals perform better than B3LYP for a model system with dispersion and ionic hydrogen-bonding interactions. J. Phys. Chem. A 117, 12590–12600 (2013).
Frisch, M. J. et al. Gaussian 09 Rev. D, Gaussian, Inc.: Walingford, CT, 2009.
Bruhn, T., Schaumloffel, A. & Hemberger, Y. SpecDis, Version 1.64; University of Würzburg (2015).
Acknowledgements
The authors are grateful for financial support from the Ministry of Science and Technology of Taiwan (MOST 103-2320-B-039-004-MY3) and King Abdullah University of Science and Technology (KAUST). Access to the Shaheen 2 High Performance Computing Facilities is acknowledged.
Author information
Authors and Affiliations
Contributions
C.-H.C. and Y.-C.W. contributed equally to the experimental design and also analyzed the data. H.-C.H. analyzed the spectroscopic data. J.-C.C. and C.-C.L. performed the biological assays. T.P.G. and K.-W.H. calculated the ECD data. S.-Y.H. collected the samples and identified the species.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chao, CH., Cheng, JC., Gonçalves, T.P. et al. Glaulactams A–C, daphniphyllum alkaloids from Daphniphyllum glaucescens. Sci Rep 8, 15417 (2018). https://doi.org/10.1038/s41598-018-33748-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33748-6
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
