
Abstract
Although recessive mutations in LAMA2 are already known to cause laminin α2-related muscular dystrophy, a rare neuromuscular disorder, large deletions or duplications within this gene are not well-characterized. In this study, we applied next-generation sequencing-based copy number variation profiling in 114 individuals clinically diagnosed with laminin α2-related muscular dystrophy, including 96 who harboured LAMA2 mutations and 34 who harboured intragenic rearrangements. In total, we detected 18 distinct LAMA2 copy number variations that have been reported only among Chinese, 10 of which are novel. The frequency of CNVs in the cohort was 19.3%. Deletion of exon 4 was detected in 10 alleles of eight patients, accounting for 27% of all copy number variations. These patients are Han Chinese and were found to have the same haplotype and sequence at the breakpoint junction, suggesting that exon 4 deletion is a founder mutation in Chinese Han and a mutation hotspot. Moreover, the data highlight our approach, a modified next-generation sequencing assay, as a robust and sensitive tool to detect LAMA2 variants; the assay identifies 85.7% of breakpoint junctions directly alongside sequence information. The method can be applied to clinical samples to determine causal variants underlying various Mendelian disorders.
Similar content being viewed by others


Introduction
Laminin α2-related muscular dystrophy (LAMA2 MD) is a rare autosomal-recessive genetic disorder affecting between 0.7 and 2.5 in 100,000 individuals in predominantly European cohorts1. It is caused by pathogenic variants in LAMA2 [MIM: 607855], which is located on chromosome 6q22-23 and consists of 65 exons2. Based on clinical features, it can be classified into two distinct entities, a severe, early-onset congenital muscular dystrophy (CMD), known as merosin deficiency or muscular dystrophy, congenital type 1A (MDC1A), which is the most frequent form of CMD, and the milder late childhood-onset limb girdle type muscular dystrophy (LGMD), known as LGMD R23 laminin α2-related3. Children with the severe form of the disease have profound hypotonia associated with muscle weakness at birth or during early infancy, poor spontaneous movements, joint contractures, and delayed motor milestones4. Unlike LGMD, these children usually do not gain independent ambulation. Specific abnormal cerebral white-matter signals are consistently observed by 1 year of age on T2-weighted MRI.
Currently, 553 unique sequence variants in LAMA2 have been reported to the Leiden Open Variation Database (accessed June 2018). Pathogenic changes include small deletions and insertions, nonsense mutations, splice site mutations, and missense substitutions. However, few large deletions or duplications have been reported5,6,7. A suspected large LAMA2 deletion, likely spanning exons 23 to 56, was initially identified by Pegoraro et al.8 bases on protein truncation test. Notably, the first fully characterized large deletion in LAMA2, a frameshift (out-of-frame) deletion of exon 56, was subsequently proven to be one of the most frequent pathogenic variants in Portuguese patients with MDC1A9. Similarly, Xiong et al.7 detected seven deletions of one or more exons in 43 Chinese patients. However, the copy-number variations (CNVs) spectrum and the characteristics of these CNVs have not been evaluated.
Genomic CNVs represent a major source of genetic diversity10. In the past decade, microarray-based profiling was introduced as a first-tier diagnostic test for genomic disorders and other diseases related to CNVs11,12. Additionally, multiplex ligation-dependent probe amplification (MLPA) enables the detection of many large deletions and duplications. However, these methods do not provide a comprehensive overview of CNVs in terms of breakpoint junctions, preventing full understanding of the pathogenic and mutational mechanisms. Although previous studies have highlighted the significance of CNVs in LAMA2 MD, diagnostic genetic testing strategies are mostly targeted at small genetic variants using, for example, next-generation sequencing (NGS)13. In addition to array-based comparative genomic hybridization (aCGH), NGS approaches can be used to detect large structural variants. Unfortunately, accurate identification of CNVs at the nucleotide sequence level by NGS remains challenging14, even though several algorithms have been developed to detect CNVs in exomes15,16,17,18 and DNA samples19,20 based on depth-of-coverage.
The aim of this study was to (1) describe the spectrum of pathogenic deletions and duplications in LAMA2 in a large cohort of patients with non-recurrent genomic rearrangements and (2) develop a modified NGS approach for LAMA2 variant detection and identification. We hope to provide a LAMA2 copy-number mutation spectrum and a diagnostic strategy for LAMA2 genetic analyses.
Materials and Methods
Editorial policies and ethical considerations
The study was reviewed and approved by the Ethics Committee of Peking University First Hospital (No. 2015[916], Beijing, China). All patients and/or their parents provided written informed consent to participate in the study and granted permission to publish medical data. Methods were compliant with the relevant guidelines and regulations.
Patient enrolment and analysis of LAMA2 mutations
In 2004–2017, 114 patients were diagnosed with LAMA2 MD at our institution. The inclusion criteria were a clinical diagnosis of muscular dystrophy characterized by muscle weakness or hypotonia with an early onset, delayed motor developmental milestones, motor-unit disease signs, a high creatine kinase level, and changes in brain white matter signals without typical structural changes observed in α-dystroglycanopathy or clinically diagnosed LGMD with typical white matter changes. Point mutations were detected by sequencing the LAMA2 gene, including all coding and flanking intronic sequences. Combined with a dosage analysis by MLPA (SALSA MLPA Kit P391-A1/P392-A1; MRC-Holland, Amsterdam, the Netherlands) and aCGH, 96 individuals were found to harbour LAMA2 mutations, including intragenic rearrangements in 34 patients from 29 families. Sufficient material for further studies was available from 29 probands, including 28 patients with deletions and 1 with a duplication. Detailed phenotypic data, including motor development, mental development, pattern of muscle involvement, joint contracture, serum creatine kinase level, brain MRI, and electromyography, obtained from all probands are listed in Table 1. DNA samples from patients and their parents were obtained from peripheral blood using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany).
Histochemistry and immunohistochemistry
Muscle biopsies were collected with informed consent from the biceps brachiis of P4, P7, P10, P18, P20, P21, and P26. Tissues were precooled with isopentane before fixation in liquid nitrogen. The histochemical staining and immunohistochemical staining were carried out and observed independently by two investigators. Laminin α2 chain was stained using 100 μL of the mouse monoclonal antibody MAB 1922 (1:5,000, 5H2, Merck Millipore, Darmstadt, Germany), and the C-terminus of dystrophin was stained with 100 μL of the mouse monoclonal antibody NCL-DYS2 (1:20, Dy8/6C5, Leica Biosystems, Newcastle, United Kingdom)6.
High-resolution aCGH analysis
High-resolution LAMA2-targeted aCGH microarrays (SurePrint G3 Microarray, 4*180K) with average probe spacing 500 base pairs were synthesized to map LAMA2 and 150 kb flanking regions. Probes were designed using the Agilent Technologies eArray tool (Santa Clara, CA, USA), and samples were tested according to the manufacturer’s recommendations. Data were analysed in Agilent Genomic Workbench version 7.0.
NGS and accurate characterization of CNVs using a composite pipeline method
Genomic DNA samples were fragmented and prepared for standard Illumina library construction. Biotinylated capture probes (MyGenostics, Beijing, China) were designed along the entire region of the LAMA2 gene (Hg19: chr6: 129204285–129837710) and sequenced using the Illumina HiSeq X Ten sequencer to obtain paired-end reads of 150 bp. Clean reads were mapped to the UCSC hg19 human reference genome using BWA. Single nucleotide polymorphisms and insertions/deletions were detected using HaplotypeCaller in GATK and functionally annotated using ANNOVAR against 1000 Genomes Project, ESP6500, dbSNP, ExAC, HGMD, and an in-house database. The pathogenicity of novel missense variants was scored using Polyphen-2, SIFT, and MutationTaster.
To predict breakpoints, structural variants were analysed in CREST according to Wang et al.21. Briefly, the soft-clipped reads were extracted from the binary alignment files, and putative breakpoints were assembled into a contig. The contig was then mapped against the reference genome (NM_000426.3) to identify candidate partner breakpoints and a match to the initial breakpoint was considered to indicate a structural variant.
Putative CNVs were identified by read-depth analysis, which is based on the ratio of reads in a test sample to reads in a control sample (Human Reference DNA mix, Promega, Madison, WI, USA). In particular, ratios less than 0.75 and above 1.25 were considered to indicate a potential deletion and duplication, respectively. CNVs of the full LAMA2 gene and its exons were calculated, and analyzed to identify approximate breakpoint positions. Finally, precise breakpoints were mapped a second time from binary alignment files in the context of breakpoints predicted in CREST.
Breakpoint-spanning long-range PCR and haplotype analysis
Long-range PCR and Sanger sequencing were performed to verify the parental derivation, and to determine sequences spanning breakpoints, as well as approximately 1,000 bp flanking each end. Primers were designed using Oligo 6.0. To analyse LAMA2 duplication by NGS, primers were designed based on the predicted tandem duplication and on head-to-tail rearrangement22. To analyse LAMA2 deletions by NGS and aCGH, primers were designed to amplify unique breakpoint junctions. Primer sequences are listed in Supplementary Table S1. Long-range PCR was performed using Takara PrimeSTAR GXL DNA Polymerase (Takara, Osaka, Japan). Variants were described according to Human Genome Variation Society guidelines for mutation nomenclature (version 2.0) and using the cDNA reference sequence (accession number NM_000426.3). Haplotype analysis was performed for P12, P13, and P14, who were heterozygous for deletion of exon 4 as previously reported7.
Analysis of mutational mechanisms underlying CNVs
RepeatMasker was used to evaluate interspersed repeat-elements at breakpoint junctions, including short interspersed nuclear elements, long interspersed nuclear elements (LINEs), long terminal repeats, DNA repeat elements, and low-complexity repeats. BLAT was used to determine the origin of sequences inserted at junctions. Blunt ends at breakpoint junctions were considered to indicate non-homologous end-joining, while microhomology was considered to indicate microhomology-mediated break-induced replication or non-homologous end-joining. Rearrangements due to Alu and long interspersed nuclear elements were identified based on the presence of such elements at breakpoint ends.
Results
Patient characteristics
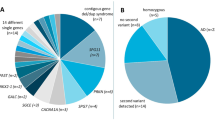
The cohort was analyzed according to Fig. 1. Clinical and neuroradiological findings are listed in Table 1 for the 29 probands with LAMA2 CNVs, of whom 27 were diagnosed with MDC1A and two were diagnosed with LGMD R23 laminin α2-related. Twenty-four probands had hypotonia and weak cry with onset at birth, while three had the same features and delayed milestones during the first 6 months. Furthermore, 24 probands never achieved independent ambulation, while five had mild muscle weakness with preserved ambulation capacity. Indeed, 13 of 96 patients genetically diagnosed with LAMA2 MD (Fig. 1) were ambulant. Three probands died of severe pneumonia at 9 years and 5 months of age. Twenty-five probands presented moderately to significantly increased creatine kinase before 2 years, which decreased after the age of 3 years and returned to physiological levels until follow-up at 6 years and older. Brain MRI showed bilateral alterations in T2 intensities in periventricular white matter after 6 months in all probands, with sparing of the corpus callosum, internal capsule, cerebellum, and brain stem. Diffuse white matter abnormalities were observed in four cases, possibly resulting in comorbid mental retardation (two patients) and epileptic seizures (two patients). Electromyography revealed myogenic damage in 23 probands, and 6 patients showed mild abnormality of peripheral nerve compound muscle action potential and nerve conduction speed. Haematoxylin and eosin staining of seven muscle biopsies showed considerable proliferation of connective and fat tissue and substantial variability in the size of muscle fibres. As assessed by immunohistochemistry of the seven biopsies, laminin α2 was weakly expressed in the one mild case (P21), but absent from the six typical cases (P4, P7, P10, P18, P20, and P26, Table 1).

Workflow of CNV analyses and breakpoint sequencing for subjects with LAMA2 MD-associated LAMA2 gene CNVs. LAMA2 gene CNVs were identified initially by MLPA assay and were further verified by high-resolution aCGH and next-generation sequencing. The sequence-based CNV structures were investigated comprehensively by CNV breakpoint sequencing.
Point mutations and novel CNVs in LAMA2
Six novel point mutations that were detected are listed in Table 2, including one nonsense mutation (c.6433A > T, p.K2145*), two frameshift mutations (c.2526_2529insACGC, p.C844Tfs*3 and c.3146del, p.G1050Afs*25), one mutation at a splice site (c.3038-7G > A), and two missense mutations (c.6584T > G, p.L2195P and c.8906G > C, p.R2969P). The missense and splice site mutations were predicted to be likely pathogenic and of uncertain significance, respectively, according to American College of Medical Genetics and Genomics classification23. Further characterization by immunohistochemistry showed complete laminin-α2 deficiency in all three patient (P4, P18, and P20).
Previously, we reported seven large deletions encompassing one or more LAMA2 exons in 43 patients with LAMA2 MD7, indicating that CNVs are relatively frequent among our clinical patients, at approximately 20%. Of the 96 cases genetically diagnosed with LAMA2 MD, 34 (35.4%) harboured heterozygous or homozygous intragenic rearrangements, such that the overall frequency of LAMA2 CNVs was 19.3% (37/192 alleles). Eight patients of Han descent (P7–P14) were homozygous or heterozygous for deletion of exon 4 from a total of 10 alleles. AACAA microhomology was observed at breakpoints in all 10 alleles, and further haplotype analysis identified a founder mutation corresponding to an in-frame deletion of 5,465 bp. Finally, immunohistochemistry showed complete laminin-α2 deficiency in P4, P7, and P10.
Heterozygous and homozygous CNVs were detected in 26 and 3 probands, respectively, and confirmed by analyzing the patients’ parents (Table 2). We identified ten novel CNVs, including nine deletions and one duplication, most (8/10, 80%) of which were predicted to cause frameshift (out-of-frame deletions). As illustrated in Fig. 1, long-range PCR was used to directly investigate six putative breakpoint junctions in three cases with exon 4 deletions. High-resolution aCGH was used to investigate 16 cases with LAMA2 MD (Supplementary. Fig. S1). Among them, 14/16 (87.5%) patients with simple CNV patterns, indicating simple genomic rearrangements, were observed. High-resolution aCGH also revealed potential CNV structural complexity in P22 (DEL-NML-DEL) that was not detected using the low-resolution MLPA. CNV was not detected by aCGH in the remaining case (P29), even though deletion of exon 63 was detected by MLPA. Further long-range PCR was used to resolve the CNV base pairs, allowing the amplification of 24/30 (80%) putative CNV breakpoint junctions. Notably, further long-range PCR was used to evaluate P17 as both junction ends overlapped with repeat elements. NGS was subsequently used to investigate the remaining 10 cases who were not analysed by aCGH, along with three cases (P17, P21, and P23) with no amplification product by long-range PCR and the one case (P29) in whom CNV was detected only by MLPA (Supplementary Fig. S2 and Table S2). Remapping resulted in the identification of a 56 bp deletion in exon 63 in P29. Of the three cases that were not resolved by long-range PCR, NGS was used to successfully resolve two, with the exception of P21, which was too large for the LAMA2-target NGS analysis coverage used in this study. All 10 cases where aCGH was not used were resolved directly using NGS data. Overall, NGS allowed direct identification of 12/14 (85.7%) putative CNV breakpoint junctions at CNVs.
Breakpoint characteristics and CNV mutational mechanisms
LAMA2 intragenic rearrangements are dispersed throughout the gene, varying in size, location, and rearrangement mechanisms (Fig. 2). Rearrangement sizes ranged from 1.3 kb to 267.1 kb, with average 71.1 kb and median 14.3 kb (Supplementary Fig. S3). Genomic rearrangements span 1 to 29 protein-coding exons, although the distribution was biased towards smaller rearrangements. In total, 71.4% (20/28) of CNVs were in the N-terminal domain (exons 1–30), especially exons 3–4. Other CNVs were found in the G domain (exons 46–63) at the C-terminus, which are believed to disrupt of the link between the extracellular matrix and dystrophin-glycoprotein24.

Global View of Identified LAMA2 Intragenic CNVs. The genomic structure of LAMA2 is presented in UCSC Genome Browser GRCh37/hg19, and custom tracks show LAMA2 intragenic CNVs. In-frame deletions were annotated with light blue color, out-of-frame deletions were annotated with dark blue color, and duplications were highlighted in red color. CNV in P21 was large and exceeded the coverage of the LAMA2-target aCGH utilized in this study, the dotted line was used to indicate the location of the uncertain breakpoint downstream.
Sequencing and alignment to the UCSC hg19 human reference genome of breakpoints at all 26 deletions (except that in P21) and 1 duplication (Table 3 and Supplementary Fig. S4) revealed simple non-recurrent rearrangements with microhomology at breakpoint junctions in 20/27 (74.0%) individuals, suggesting double-strand DNA breaks followed by microhomology-mediated end-joining. In 5/27 (18.5%) rearrangements, an insertion of 1–328 bp was found between breakpoints. One of these insertions (in P22) was most likely mediated by fork stalling and template switching/microhomology-mediated break-induced replication, and one, a short stretch of 28 bp in P6, was mapped to the reference sequence close to the breakpoint, suggesting serial replication stalling and re-replication. CNVs in P4, P23, and P24 contained short insertions with random nucleotides (<5 bp) at breakpoints, suggesting replication-independent non-homologous end-joining. Only breakpoint junctions in P5 and P25 consisted of two blunt ends without insertions. Finally, the duplication was confirmed to be a tandem duplication at the LAMA2 locus.
Using RepeatMasker in the UCSC Genome Browser, 96 Alu and 83 partial L1 repetitive elements longer than 100 bp were detected throughout the LAMA2 non-coding region (Supplementary Fig. S5). Indeed, 11 of 27 (40.7%) junction ends overlapped with at least one type of repeat element, which is much lower than in the DMD gene25,26, but higher than the average frequency (30.5%) of repetitive sequences in LAMA2. Only the rearrangement in P17 is likely to be due to L1-mediated non-allelic homologous recombination, with a 471 bp region of identity. Alu elements were not involved in genomic rearrangements in our cohort. A high frequency of single- or oligonucleotide changes close to the breakpoints was detected in the LAMA2-associated CNVs analyzed in this study (Table 2), all of which are absent from 1000 Genomes Project and ExAC27, consistent with error-prone replicative repair mechanism in CNV mutagenesis28.
Correlation between CNV contents and disease severity
The length of deletions and the corresponding exons that are deleted may partially account for the spectrum of LAMA2 MD phenotypes and severity. P29, who does not harbour CNVs, was excluded from the following analysis of genotype-phenotype correlation. The remaining 28 probands were categorized into two groups according to phenotype. In a comparison of LAMA2-associated CNV sizes, the deleted genomic segments were considerably longer in the LGMD R23 laminin α2-related group (P21, P28) than in the MDC1A group. Both deletions in the LGMD R23 laminin α2-related group were located in the G domain at the C-terminus. One half of the LAMA2 deletions (14/28, 50%) were predicted to induce frameshift truncations (out-of-frame deletions; Fig. 2), all in-frame deletions were found in MDC1A group in combination with a second truncating frameshift or a nonsense mutation, except in three patients with missense mutations instead. Nevertheless, these three patients (P4, P13, and P27) had typical MDC1A phenotypes. Strikingly, individuals with the same CNV may have different maximal motor milestones. For example, P13 carried the Chinese founder mutation, with a deletion of exon 4, but was independently ambulant, possibly due to compound heterozygosity with a missense mutation.
Discussion
In this study, 34 cases from 29 families were genetically characterized in detail for pathogenic CNVs in LAMA2, following detection of non-recurrent genomic rearrangements among a large cohort of patients with LAMA2 MD. Moreover, a modified NGS assay was developed to detect and identify LAMA2 variants. Deletion of exon 4 was detected in eight non-consanguineous Chinese Han patients with MDC1A (10/37 disease alleles, accounting for 27% of all CNVs). Breakpoint analysis revealed AACAA microhomology at the junction in all 10 disease alleles, and haplotype analysis identified a founder mutation consisting of an in-frame deletion of 5,465 bp. Therefore, screening for LAMA2 point mutations, followed by analysis of LAMA2 CNVs, especially exon 4 deletion, may be appropriate as an initial strategy for patients with features consistent with congenital muscular dystrophy, such as muscular dystrophy combined with white matter changes in brain MRI.
Based on the two largest studies of LAMA2 MD patients to date, we estimated that the overall frequency of CNVs may be as high as 18.6% (55/296 alleles), highlight the importance of screening for CNVs in suspected cases. These alleles consist of 37/192 alleles in our cohort, including from 96 patients with LAMA2 mutations (Xiong et al.7 and this study), and of 18/104 alleles analyzed by Oliveira et al.5 in 52 patients. Our data also support the existence of two possible hotspots for large LAMA2 deletions, one at exons 3 and 4 and another at exons 56 to 65 at the 3′ region of the gene, as first described by Oliveira et al.5. Strikingly, we detected 27 intragenic LAMA2 deletions but only one intragenic duplication in our cohort. Indeed, only one other pathogenic heterozygous in-frame duplication of exons 28 and 29 has been documented previously5, and which is different from the novel duplication encompassing exons 5 to 8 in patient P16.
Currently, genomic sequencing is the main strategy to diagnose pathogenic LAMA2 variants, with the highest sensitivity of approximately 80%. aCGH and MLPA are frequently used to detect CNVs as well, although these methods do not resolve breakpoint junctions and genomic orientation. For example, aCGH may generate spurious calls due to non-biological hybridization signals. Indeed, patient P7 was diagnosed by aCGH as carrying a small deletion, but mapping endpoints by specific PCR and sequencing instead identified the arrangement as a founder mutation involving exon 4. Similarly, samples from P23 produced significantly more calls than average, and mapping subsequently failed due to high levels of noise. On the other hand, data obtained by MLPA were limited as well. The probe mixes P391 and P392 contain one or more probes for all LAMA2 exons except 18, 44, and 48, therefore, CNVs at these sites may have been undetectable. We note that the false-positive rate for MLPA has been determined previously, along with sequence alterations that may compromise probe affinity. Several algorithms have been developed to identify CNVs from depth of coverage. However, the newest whole-exome sequencing platforms detect only deletions of three exons or larger, while smaller events are not reliably detected29,30. Whole genome sequencing has also been used as a standalone assay to detect genetic variants31, but this approach cannot detect CNVs shorter than 1 kb and may miss some longer CNVs as well. Our results show that more than half of LAMA2 intragenic CNVs (53.4%, 15/28) span just one exon, the smallest 56 bp, highlighting the sensitivity of our method for CNV analysis. Thus, our custom-designed NGS approach may delineate large genomic rearrangements in addition to sequence variations, with high accuracy and specificity as well as reasonable cost and practicality.
Although several mechanisms have been proposed to drive genomic rearrangements28, we found that replication-based mechanisms such as fork stalling and template switching/microhomology-mediated break-induced replication may account for most LAMA2 intragenic CNVs. In this study, we identified blunt ends at two breakpoint junctions and random nucleotide insertions <5 bp at three breakpoints, both features consistent with “information scars”32,33 that are typically formed by replication-independent non-homologous end-joining. Twenty simple non-recurrent rearrangements showed microhomology at the breakpoint junctions, also indicating non-homologous end-joining or fork stalling and template switching/microhomology-mediated break-induced replication34. Additionally, serial replication stalling was likely in one case, further indicating that replication-based mechanisms contribute to LAMA2 intragenic CNVs. Indeed, high levels of microhomology at breakpoint junctions indicate replication-based mechanisms35,36, the significance of which may have been previously underestimated.
Mammalian replicons span 75–150 kb on average, and human genes are 27 kb on average37. Accordingly, the large size of LAMA2, as well as a larger number of intragenic replication origins, may explain the high frequency of intragenic CNVs. Using RepeatMasker, we found that 40.7% (11/27) of breakpoint junctions contained one or more repeat elements, a frequency higher than the average percentage of repeat elements in the entire LAMA2 gene. Additionally, LINE elements are most likely than SINE elements to mediate gene rearrangements in LAMA2. For example, the upstream breakpoints in P26–P28 are fragments of L3 (chr6: 129816155–129816735), resulting in deletion of exon 59–63 in three unrelated individuals with the same breakpoint. Recurrence of this deletion may be due to a founder effect, but further studies are needed to validate this hypothesis. Additionally, 4/11 instances of rearrangements are clustered at two LINE elements at introns 9 and 12, which are thus potential intragenic-rearrangement hotspots.
In summary, we provide for the first time a novel perspective on the spectrum of CNVs in LAMA2. In particular, we demonstrate that deletion of exon 4 is a founder mutation in Chinese Han population and the exon itself being a mutation hotspot. Moreover, we describe a novel NGS approach to detect and sequence CNV breakpoints. Our locus-centered analysis provides valuable insight into the molecular aetiology of LAMA2 MD, and may help clinicians provide accurate and reliable genetic counseling, prenatal diagnosis, and gene therapy for those at risk.
Web resources
The URLs for data presented herein are as follows:
Leiden Open Variation Database | |
BWA | |
HaplotypeCaller of GATK | |
ANNOVAR | |
Polyphen-2 | |
SIFT | |
Mutation Taster | |
Oligo |
References
Quijano-Roy, S., Sparks, S. E. & Rutkowski, A. GeneReviews® [Internet]. LAMA2-Related Muscular Dystrophy. Seattle: University of Washington, 1993–2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK97333/.
Helbling-Leclerc, A. et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet 11, 216–218 (1995).
Volker, S., Alexander, M. & Bjarne, U. On behalf of the 229th ENMC workshop study group, creat229th ENMC international workshop: Limb girdle muscular dystrophies-nomenclature and reformed classification, 17–19 March 2017, Naarden, The Netherlands, Neuromuscular Disord, https://doi.org/10.1016/j.nmd.2018.05.007 (2018).
Philpot, J., Sewry, C., Pennock, J. & Dubowitz, V. Clinical phenotype in congenital muscular dystrophy: Correlation with expression of merosin in skeletal muscle. Neuromuscul Disord 5, 301–305 (1995).
Oliveira, J. et al. Reviewing Large LAMA2 Deletions and Duplications in Congenital Muscular Dystrophy Patients. J Neuromuscul Dis 1, 169–179 (2014).
Ding, J. et al. Clinical and molecular genetic analysis of a family with late-onset LAMA2-related muscular dystrophy. Brain Dev 38, 242–249 (2016).
Xiong, H. et al. Genotype/phenotype analysis in Chinese laminin-α2 deficient congenital muscular dystrophy patients. Clin Genet 87, 233–243 (2015).
Pegoraro, E. et al. Laminin alpha2 muscular dystrophy: Genotype/phenotype studies of 22 patients. Neurology 51, 101-110 (1998).
Oliveira, J. et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet 74, 502–512 (2008).
Zhang, F., Gu, W., Hurles, M. E. & Lupski, J. R. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet 10, 451–481, https://doi.org/10.1146/annurev.genom.9.081307.164217 (2009).
Tang, Y. C. & Amon, A. Gene copy-number alterations: a cost-benefit analysis. Cell 152, 394–405 (2013).
Aradhya, S. et al. Exon-level array CGH in a large clinical cohort demonstrates increased sensitivity of diagnostic testing for Mendelian disorders. Genet Med 14, 594–603 (2012).
Bönnemann, C. G. et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord 24, 289–311 (2014).
Lek, M. & MacArthur, D. The challenge of next generation sequencing in the context of neuromuscular diseases. J Neuromuscul Dis 1, 135–149 (2014).
Dong, Z. et al. Low-pass whole-genome sequencing in clinical cytogenetics: a validated approach. Genet Med 18, 940–948 (2016).
D’Aurizio, R. et al. Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2. Nucleic Acids Res 44, e154 (2016).
Gambin, T. et al. Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res 45, 1633–1648 (2017).
Pfundt, R. et al. Detection of clinically relevant copy-number variants by exome sequencing in a large cohort of genetic disorders. Genet Med 19, 667–675 (2017).
Liu, S. et al. Traditional karyotyping vs copy number variation sequencing for detection of chromosomal abnormalities associated with spontaneous miscarriage. Ultrasound Obstet Gynecol 46, 472–477 (2015).
Fu, X. et al. Novel copy number variation of POMGNT1 associated with muscle-eye-brain disease detected by next-generation sequencing. Sci Rep 7, 7056 (2017).
Wang, Y. et al. Characterization of 26 deletion CNVs reveals the frequent occurrence of micro-mutations within the breakpoint-flanking regions and frequent repair of double-strand breaks by templated insertions derived from remote genomic regions. Hum Genet 134, 589–603 (2015).
Lee, J. A., Carvalho, C. M. & Lupski, J. R. A. DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131, 1235–1247 (2007).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424 (2015).
Yurchenco, P. D., McKee, K. K., Reinhard, J. R. & Rüegg, M. A. Laminin-deficient muscular dystrophy: Molecular pathogenesis and structural repair strategies. Matrix Biol 17, 30333–30335, https://doi.org/10.1016/j.matbio.2017.11.009 (2017).
Oshima, J. et al. Regional genomic instability predisposes to complex dystrophin gene rearrangements. Hum Genet 126, 411–423 (2009).
Chen, C. et al. Screening of Duchenne muscular dystrophy (DMD) mutations and investigating its mutational mechanism in Chinese patients. Plos One 9, e108038 (2014).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Carvalho, C. M. & Lupski, J. R. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17, 224–238 (2016).
Retterer, K. et al. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet Med 17, 623–629 (2015).
Wang, J. et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods 8, 652–654 (2011).
Trost, B. et al. A Comprehensive Workflow for Read Depth-Based Identification of Copy-Number Variation from Whole-Genome Sequence Data. Am J Hum Genet 102, 142–155 (2018).
Gu, W., Zhang, F. & Lupski, J. R. Mechanisms for human genomic rearrangements. Pathogenetics 1, 4 (2008).
Hsiao, M. C. et al. Decoding NF1 Intragenic Copy-Number Variations. Am J Hum Genet 97, 238–249 (2015).
Hastings, P. J., Ira, G. & Lupski, J. R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5, e1000327 (2009).
Ottaviani, D., LeCain, M. & Sheer, D. The role of microhomology in genomic structural variation. Trends Genet 30, 85–94 (2014).
Zhang, L. et al. Efficient CNV breakpoint analysis reveals unexpected structural complexity and correlation of dosage-sensitive genes with clinical severity in genomic disorders. Hum Mol Genet 26, 1927–1941 (2017).
Venter, J. C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001).
Pegoraro, E., Fanin, M., Trevisan, C. P., Angelini, C. & Hoffman, E. P. A novel laminin alpha2 isoform in severe laminin alpha2 deficient congenital muscular dystrophy. Neurology 55, 1128–1134 (2000).
Beytia Mde, L. et al. High creatine kinase levels and white matter changes: clinical and genetic spectrum of congenital muscular dystrophies with laminin alpha-2 deficiency. Mol Cell Probes 28, 118–122 (2014).
He, Y. et al. Congenital muscular dystrophy with primary partial laminin alpha2 chain deficiency: molecular study. Neurology 57, 1319–1322 (2001).
Geranmayeh, F. et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 20, 241–250 (2010).
Kevelam, S. H., van, Engelen, B. G., van, Berkel, C. G., Küsters, B. & van, der, Knaap, M. S. LAMA2 mutations in adult-onset muscular dystrophy with leukoencephalopathy. Muscle Nerve 49, 616–617 (2014).
Valencia, C. A. et al. Comprehensive mutation analysis for congenital muscular dystrophy: a clinical PCR-based enrichment and next-generation sequencing panel. Plos One 8, e53083 (2013).
Acknowledgements
This work was supported by grants from The National Key Research and Development Program of China (No. 2016YFC0901505); National Natural Science Foundation of China (No. 81571220); Beijing key laboratory of molecular diagnosis and study on pediatric genetic diseases (No. Z141107004414036, BZ0317); Beijing Municipal Science and Technology Commission (No. Z151100003915126). The authors are grateful to all the doctors, patients and their families who have participated in this study and thank Dr. Monkol Lek, PhD, for his comments and criticisms on the manuscript.
Author information
Authors and Affiliations
Contributions
Study conceptualization: L.G., F.Z., Y.W.J., X.R.W., H.X. Data collection: L.G., A.J.L., J.D., B.M., Y.H., X.L.Z., D.D.T., H.P.Y., X.N.F., Y.B.F. Data analysis: L.G., K.G., R.Q.D., L.Z., S.J.S., J.W. Wrote manuscript: L.G., H.X. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ge, L., Liu, A., Gao, K. et al. Deletion of exon 4 in LAMA2 is the most frequent mutation in Chinese patients with laminin α2-related muscular dystrophy. Sci Rep 8, 14989 (2018). https://doi.org/10.1038/s41598-018-33098-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33098-3
Keywords
This article is cited by
-
Early diagnosis of congenital muscular pathologies using next-generation sequencing: experiences from a tertiary center in Morocco
Egyptian Journal of Medical Human Genetics (2023)
-
Comprehensive analysis of immune-related lncRNAs and their clinical relevance in gastric adenocarcinoma
Functional & Integrative Genomics (2023)
-
Diagnostic interest of whole-body MRI in early- and late-onset LAMA2 muscular dystrophies: a large international cohort
Journal of Neurology (2022)
-
Natural history and genetic study of LAMA2-related muscular dystrophy in a large Chinese cohort
Orphanet Journal of Rare Diseases (2021)
-
Fat mass- and obesity-associated (FTO) gene promoted myoblast differentiation through the focal adhesion pathway in chicken
3 Biotech (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
