
Abstract
Several members of the EPH kinase family and their ligands are involved in blood pressure regulation, and such regulation is often sex- or sex hormone-dependent, based on animal and human genetic studies. EPHB6 gene knockout (KO) in mice leads to hypertension in castrated males but not in un-manipulated KO males or females. To assess whether this finding in mice is relevant to human hypertension, we conducted a human genetic study for the association of EPHB6 and its two ligands, EFNB1 and EFNB3, with hypertension in hypogonadic patients. Seven hundred and fifty hypertensive and 750 normotensive Han Chinese patients, all of whom were hypogonadic, were genotyped for single nucleotide polymorphisms (SNPs) within the regions of the genes, plus an additional 50 kb 5′ of the genes for EPHB6, EFNB1 and EFNB3. An imputed insertion/deletion polymorphism, rs35530071, was found to be associated with hypertension at p-values below the Bonferroni-corrected significance level of 0.0024. This marker is located 5′ upstream of the EFNB3 gene start site. Previous animal studies showed that while male EFNB3 gene knockout mice were normotensive, castration of these mice resulted in hypertension, corroborating the results of the human genetic study. Considering the significant associations of EFNB3 SNPs with hypertension in hypogonadic males and supporting evidence from castrated EFNB3 KO mice, we conclude that loss-of-function variants of molecules in the EPHB6 signaling pathway in the presence of testosterone are protective against hypertension in humans.
Similar content being viewed by others

Introduction
EPH kinases are receptor tyrosine kinases. They are divided into A and B subfamilies according to sequence homology1. EPH ligands are cell surface molecules ephrins (EFN), which are also divided into A and B subfamilies, according to how they anchor on the cell surface (glycosylphosphatidylinositol anchoring for EFNAs and transmembrane anchoring for EFNBs)1,2. The interactions between EPHs and EFNs are promiscuous, although in general EPHA subfamily members preferably interact with EFNAs, and EPHBs interact with EFNBs1,2.
EPHs and EFNs are critical in the development and function of the central nervous system3, immune system4,5,6,7,8,9,10,11,12,13,14,15,16, and digestive system17, and in different tissues and processes, such as bone homeostasis18,19, angiogenesis20, etc.20,21,22,23.
We have recently reported that EPHB6, EPHB4, EFNB1, EFNB2 and EFNB3 are novel regulators in blood pressure (BP)24,25,26,27,28. While deletion of EPHB6, EFNB1 and EFNB3 results in increased BP24,25,26, EPHB4 and EFNB2 deletion leads to lower BP27,28 in mice. Such BP regulation is often sex- and/or sex hormone-specific24,25,26,27,28. Thus, these molecules likely function as yin and yang in maintaining BP homeostasis, with sex hormones as modifiers. Our human genetic studies corroborate the findings in mice. We have found that five single nucleotide polymorphisms (SNPs) in the 3′ region of EFNB2 gene are significantly associated with hypertension for male but not female patients with type 2 diabetes, and the coding (minor) allele of these SNPs are protective against hypertension in male Caucasians28. In the same cohort, two SNPs in the 3′ region of EFNB3 gene are associated with hypertension risks in male but not female Caucasians29. In a separate cohort of International Blood Pressure Consortium, we have revealed that a SNP in GRIP1 gene, whose protein product is in the EFNB3 signaling pathway, is significantly associated with diastolic BP of 69,395 individuals30. Taken together, these mouse and human studies confirm that EPHs and EFNs are previously unknown BP regulators, and they deserve our attention in our effort to control the epidemic health issue of hypertension.
In our EPHB6 KO mouse studies, EPHB6 is deleted in all tissues8,24. In these mice, male KO presented normal BP, but after castration, they become hypertensive24. We have revealed that the vascular smooth muscle cells (VSMCs) and adrenal gland chromaffin cells (AGCCs) are EPHB6 target cells for this BP phenotype24. The default function of EPHB6 in VSMCs is to decrease their contractility. Therefore, when EPHB6 is deleted, the opposite will occur. However, in spite of the increased VSMC contractility, the BP in the male KO mice remains normal because EPHB6 KO in AGCCs also causes reduced ambient catecholamine secretion24, which neutralizes the effect of VSMC contractility increase. However, castration elevates the AGCC catecholamine secretion to a normal level24. This, in conjunction with high VSMC contractility in the KO mice, leads to overt hypertension in the castrated KO mice24. This raises an interesting hypothesis relevant to human hypertension: for a subpopulation of males with loss-of-function mutations in the EPHB6 gene or genes in the EPHB6 signaling pathways, they might enjoy protection against hypertension; however, if they suffer from hypogonadism at the same time, they might become hypertensive due to detrimental effects of other genetic or environmental factors.
We conducted a human genetic study to assess this hypothesis. Seven hundred and fifty hypertensive patients with hypogonadism were used as cases, and 750 normotensive hypogonadic patients as controls. SNPs in the EPHB6, EFNB1 and EFNB3 genes plus 50-kb 5′ upstream sequences of these genes were analyzed by Illumina GoldenGate custom-made arrays, protein products EFNB1 and EFNB3 being the ligands of EPHB6. The results show that the one SNP in the 5′ end upstream of the EFNB3 gene start site was significantly associated with hypertension risks.
Patients, Materials and Methods
Patient population
A total of 4,480 male patients ≥40 years old from the Cardiology Ward, Endocrinology Ward, and Physical Examination Center of First Affiliated Hospital, College of Medicine, Zhejiang University in Hangzhou, China, were recruited for this study. They were tested for total plasma testosterone levels. Those with hypogonadism (plasma total testosterone levels <346 ng/dL, the cut-off level recommended by the International Society for the Study of the Ageing Male31 were retained. Among those hypogonadic individuals, 982 were diagnosed with primary hypertension and were considered as cases. The primary hypertension phenotype was defined as having a measurement of systolic pressure >140 mm Hg or diastolic pressure >90 mmHg, or having been actively treated for hypertension, excluding known conditions or medication that could cause BP increase. All other medical conditions were allowed as long as they were not the cause of hypertension. All types of medications in the last three months were allowed except those that are known to affect testosterone levels, such as testosterone replacement therapy. Seven hundred and eighty eight normotensive subjects were considered as controls. Seven hundred and fifty cases and 750 controls were selected with an attempt to match their ages as closely as possible. The means ± SE (standard error) of systolic and diastolic BP of the case and control groups are listed in Table 1. Differences of the case and control groups in potential covariate phenotype parameters (such as age, plasma testosterone level, heart rates, body mass index (BMI), serum uric acid levels and smoking status) that could be implicated in causing hypertension are listed in Table 2.
Ethics statement
This human genetic study was approved by the Ethics Committee of the First Affiliated Hospital of Zhejiang University (No. 2013-145), and was carried out in accordance with guidelines of the Committee. Informed consent was obtained from all the subjects recruited in this study. All experiments were conducted in accordance with other relevance guidelines and regulations of the local government.
Blood sample collection and plasma total testosterone measurements
Venous blood samples were drawn from all subjects after an overnight fast of at least eight hours. Five mL of blood was collected into vacuum tubes with the anticoagulant EDTA-K+ and centrifuged at the site of collection within one hour. Cell pellets were frozen until DNA extraction.
Plasma total testosterone levels were measured with Siemens Immulite 2000 Total Testosterone Kits on Siemens Immulite 2000 Immunoassay Analyzer according to the manufacturer’s protocols.
DNA extraction and purification
Sample DNA was extracted using DNeasy Blood & Tissue Kit (Cat. 69506, QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Purified DNA quantity and quality were assessed by Qubit®2.0 Fluorometer (Q32866, Invitrogen, Carlsbad CA, USA) and 1% agarose gel electrophoresis. Samples with DNA quantity ≥2 µg and optical density (OD) 260/280 = 1.8–2.0 were submitted to SNP assay.
Candidate tag SNPs
Chromosomal regions containing sequences from the candidate genes EPHB6, EFNB1 and EFNB3 were identified from the UCSC Genome Browser32 using Build 37/hg19 Human Genome Assembly, and their SNPs were used for genotyping or imputation. An additional 50 kb of DNA sequence 5′ upstream of each gene were also included in order to examine potential gene regulatory elements. Tag SNPs within these regions were chosen using the Tagger program33 with the Han Chinese in Beijing (CHB) linkage-disequilibrium (LD) data. For EPHB6, tag SNPs in the first 2.4 kb of the EPHB6 gene between positions 142552792 and 142555251, which ended in the first exon, plus 50-kb 5′ sequence was selected for genotyping. Tag SNPs were chosen with to represent proxy SNPs with a minimum linkage disequilibrium (LD) value coefficient of determination (r2) >0.80 and minor allele frequency >0.05. Candidate tag SNPs were then submitted for analysis by Illumina Software for compatibility with the GoldenGate multiplexing process. Alternative Tag SNPs were chosen for those Tag SNPs determined to be incompatible with the multiplexing technology and the new Tag SNPs were then re-submitted for a new Illumina software analysis to determine the compatibility of the new set of SNPs including previously compatible SNPs and the new alternative Tag SNPs. This process continued in an iterative manner until all Tag SNPs were found to be compatible for use by the multiplexing technology. Ultimately, seven tag SNPs were chosen for EPHB6, seven tag SNPs for EFNB1, and nine tag SNPs for EFNB3 (Table 3). Their chromosomal positions and minor allele frequencies are also presented (Table 3). Bonferroni-corrected critical significance level (pcrit) were calculated for SNP and hypertension associations assuming tag SNPs represent independent statistical tests when performing association analysis across the regions: pcrit = 0.0071 for EPHB6 alone (pcrit = 0.05/7); pcrit = 0.0071 for EFNB1 alone (pcrit = 0.05/7); pcrit = 0.0056 for EFNB3 alone (pcrit = 0.05/9), and pcrit = 0.0024 for the experiment-wide analysis of all the three genes combined (pcrit = 0.05/21).
Tag SNP genotyping
The tag SNPs were genotyped by the Shanghai Biotechnology Corporation using the Illumina GoldenGate genotyping platform according to the manufactures instructions. Those SNPs with a call rate less than 90% were filtered out and not analyzed.
Association analysis
The genotyped SNPs were tested for Hardy-Weinberg equilibrium and were then used to impute additional SNPs within the region covered by the tag SNPs using the IMPUTE2 program34 and the 1000 Genomes Phase 3 East Asian LD dataset35. Thirty-eight additional SNPs were imputed for the EFNB1 region, 124 for the EFNB3 region and 204 for the EPHB6 region. The imputed and the genotyped tag SNPs were then analyzed for association with hypertensive versus normotensive status across all three regions employing the PLINK36 program, and using a logistic regression model with or without covariates. Each SNP was assigned a reference allele and an alternative allele. The number of reference alleles in each individual was entered as its genotype value. These were summed over all cases and controls and entered into the logistic regression equation as the genotype term. This is analogous to an additive genetic model. Therefore, odds ratios refer to the reference allele which is noted in the tables.
For the X-linked EFNB1 gene, the SNP imputation also utilized IMPUTE2, which handles imputation for both sex and autosomal chromosomes. The program essentially has two steps. In the first step the haplotypes are estimated from the genotype values and allele frequencies using a Bayesian expectation-maximization iterative procedure. The second step determines the alleles of non-genotyped SNPs based upon their occurrence on the estimated haplotypes as determined by the 1000 genome project data for Chinese Asian populations. The haplotype determination step will have a small error associated with it for autosomal and for X-linked genes in females. However, males are hemizygous for X-linked genes, so the genotype sequences in males are the haplotypes. IMPUTE2 treats X-linked genes in males as homozygotes for the purpose of haplotype estimation, so for males (the patients in our cohort are all males), there is no error in the haplotype estimation step. Hence, X-linked haplotype determination is error-free in our case, and is thus more accurate. However, for the X-linked genes in males, there is only half the number of alleles to count for the association analysis compared to autosomal genes. As a consequence, we have a smaller sample size for X-linked SNPs compared to autosomal SNPs, and there will be higher p-values for an association of the same effect size.
Results
In this study, we first screened 4,480 male patients, and retained only those with hypogonadism, diagnosed according to the criteria of the International Society for the Study of Ageing Males (plasma testosterone levels <346 ng/dL). It is to be noted that although the mean BP (systolic as well as diastolic) was higher in the cases than the controls as expected (Table 1), they did not reach the hypertension diagnostic criteria, i.e., systolic pressure >140 mmHg and/or diastolic pressure >90 mmHg. This is because the majority of the cases (93.7%) were previously diagnosed and were under anti-hypertension medication; their BP was largely under control to be within the normal range.
Means or percentages of various additional parameters, age, plasma testosterone levels, heart rates, body mass index (BMI), serum uric acid levels and smoking status that might affect hypertension status are presented in Table 2. Simple logistic regression of each of these individual parameters against case versus control status was conducted to assess the significance of the differences in these parameters between cases and controls. The p-values of these tests are shown in Table 2. Despite our efforts to match the ages of the cases and controls, there was still a slight but statistically significant difference in mean age (p = 0.0104). The plasma testosterone levels of the cases were also slightly lower than those of the controls, but the difference was not significant. The heart rates of the cases were significantly slower (p = 0.0018) than those of the controls. This is likely due to reflexive feedback regulation of the higher BP in the cases. The cases had significantly higher BMI than the controls (p = 1.91 × 10−12). The higher BMI could be a contributing factor to hypertension in the cases. Finally, the cases had significantly higher serum uric acid levels than the controls (p = 0.0002). This probably reflects kidney damage caused by hypertension, because we only recruited patients with primary hypertension, and none of the cases was hypertensive due to primary renal diseases.
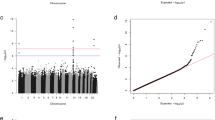
The potential covariate parameters listed in Table 2 were used in a stepwise multiple logistic regression against case and control status. The results (Table 4) show that all of these parameters, when combined in a multiple regression model, were significantly different between cases and controls, except testosterone levels, which were nearly significant with a p-value slightly above 0.05 (p = 0.0506), and smoking status, which was not significant. We thus used the significant and near-significant parameters, i.e., age, testosterone level, heart rate, BMI and serum uric acid levels, as covariates in our following genetic association analysis in order to eliminate the possibility of confounding them with genotype association to hypertension, even though we consider that some of them might be consequences rather than causes of hypertension. The genetic association was tested using logistic regression models with or without the covariates. The results of the association study of all genotyped tag SNPs and imputed SNPs across all three tested gene regions (EPHB6, EFNB1, and EFNB3) are presented in the supplementary materials (Supplementary Table 1). Reference alleles used in coding the genotypes, alternative alleles, minor alleles, minor allele frequencies, information scores for the imputed SNPs, odds ratios of association with respect to the reference allele, standard error of odds ratios and p-values of association models with or without covariates are presented for each SNP. Locus zoom plots for association across the three gene regions are presented in Figure 1 for both models with and without covariates.

LocusZoom plots for association of all SNPs assayed across EPHB6, EFNB1 and EFNB3 gene regions for regression models with and without covariates. Left Y-axis: -Log10 (p-value) of the SNP association with hypertension. Right Y-axis: Recombination rate (cM/Mb). X-axis: position of SNPs and EFNB3 gene in chromosome 7. Color bar: r2 value.
In general, the p-values of association for the covariate models displayed similar general patterns of association but were slightly lower than in the models that did not incorporate the covariates for many SNPs. This suggests that the effects of covariates and genotypes were somewhat but not completely independent. No SNPs approached significance in the tested EPHB6 or EFNB1 gene regions in models with or without covariates; however, one imputed SNP located 5′ of ENFB3, rs35530071 (Chromosome 17 position 7563124 Build 37/hg 19, p = 0.00087 for the covariate model and p = 0.00170 for the model without covariates; Table 5) reached 3-gene experiment-wide significance (pcrit ≤ 0.0024). Two other nearby imputed SNPs, rs73246870 and rs61209339 (Chromosome 17 positions 7559777 and 7560279) reached significance for the Bonferroni correction for the single gene-only analysis (9 tag SNPs pcrit = 0.0055). The most significant nearby genotyped SNP was rs1050540 (Chromosome 17 positon 7560742, p = 0.0099 for the covariate model and p = 0.01663 for the model without covariates), which did not reach Bonferroni-corrected significance level.
These 4 SNPs are all located in the far 5′ region upstream of the start site for EFNB3. SNP associations with hypertension within this region are shown in Table 5.
To assess the impact of each of the covariates on the significant SNP associations with hypertension, we re-analyzed the multiple logistic regression associations for the above-mentioned four SNPs using the R software37, which provides the estimates and p-values of significance for all covariate terms as well as for the genotype terms. These values are presented in Table 6. The blood testosterone term had a negative coefficient indicating that it is negatively correlated with hypertension, as expected. It became slightly more significant when the multiple regression model included the genotype terms, the p-values going from just above 0.05 without a genotype term (Table 4) to near 0.041–0.045 (Table 6). The general orders of magnitude of significance remained the same for the other covariates for models with and without the genotype term, with BMI being the most significant covariate.
Discussion
Our previous animal study demonstrates that castrated EPHB6 KO mice exhibit hypertension, suggesting that EPHB6 and/or its signalling pathways are involved in testosterone-dependent blood pressure control. To assess the relevance of this phenotype to human hypertension, in this study we investigated the association of SNPs in the EPHB6 gene, and in the genes of its two ligands, EFNB1 and EFNB3, with hypertension in Han Chinese hypogonadic males.
EPHB6 has three potential ligands, i.e., EFNB1, EFNB2 and EFNB3. The deletion of EPHB6, EFNB1 and EFNB3 all result in hypertension in mice, while the deletion of EFNB2 leads to an opposite phenotype, i.e., hypotension. Thus, we speculated that EFNB1 and EFNB3 are more relevant to EPHB6 signalling for the hypertension phenotype, and hence they were included in our current hypertension association study. Regulatory elements (enhancers or repressors) are often present in the 5′ region of genes. So SNPs in the 50-kb regions upstream of these genes were added for the analysis. While no SNPs in analyzed EPHB6 or EFNB1 regions were significantly associated with hypertension, three markers (one at a 3-gene experiment-wide significance level, and two at an EFNB3 single-gene significance level) located 5′ upstream of the EFNB3 gene start site were significantly associated with hypertension in this population even after correction for all of the covariates, which were also all significantly or near-significantly associated with hypertension. A quantitative analysis of the association of the SNPs of these three gene regions with the BP levels, after adjusting for medication (by adding 15 mmHg to SP and 10 mmHg to DP), showed similar results (data now shown) as the qualitative analysis.
Why were SNPs in the EFNB3 gene but not the EPHB6 gene significantly associated with hypertension in hypogonadic males, in spite of the EPHB6 KO leading to hypertension in castrated mice? In mice, when we delete EPHB6, its signalling, including the forward signalling from EFNBs to EPHB6, and reverse signalling from EPHB6 to EFNBs, could be compromised. Any defect in these signalling pathways could lead to the hypertension phenotype, while EPHB6 itself might not be critically important. Indeed, EFNB3 deletion also causes hypertension26, suggesting that at least some BP-related phenotype in EPHB6 KO mice is involving EFNB3 via signalling.
Another possible explanations are that (1) significantly associated SNPs might lie in the remaining 13.6 kb of the EPHB6 gene or even in the 3′ flanking sequence of EPHB6 gene, which are yet to be tested; or (2) while the lack of EPHB6 function leads to hypertension as demonstrated by the gene knockout results in the mouse model, it may simply be that there are no functional polymorphisms present within the analysed EPHB6 gene region in this human population that could result in loss of function of the gene.
We did a search of eQTL of the three most significant SNPs in the EFNB3 gene region, i,e., rs35530071 (located at 45,396 bp 5′ upstream of the EFNB3 gene start site), SNP rs73246870 (located 48,743 bp 5′ upstream of EFNB3 gene start site) and rs61209339 (located 48,241 bp 5′ upstream of EFNB3 gene start site, using GRex38. The database did not show any significant association of these SNPs with EFNB3 expression changes in any tissue, suggesting that these SNPs are unlikely functional mutations, or such expression data are not yet available. These SNPs appear to be in high LD with each other, and thus indicate the presence of a single functional polymorphism within this LD block located from 48.5 to 37.9 kb 5′ upstream of the EFNB3 gene. It is not uncommon to have regulatory elements located at such a distance 5′ to the start site of the gene. For example, β-globin has an enhancer at 50-kb 5′ upstream of its start site39; apoB has an effective enhancer at 55-kb 5′ upstream of its start site40. Several genes (e.g., FOXG1 and MYC) even have enhancers at a distance 1 mb or farther from the start sites41. In some cases, there are additional genes located between an enhancer at a large distance away and the gene it regulates. Yet the enhancer would ignore the closest promoters and favour the one situated at a distance42. In the nuclei, DNA is coiled, and thus an enhancer at a seemingly long linear distance from a promoter in a three-dimensional space might actually be at the proximity of the latter, due to directed or assisted looping of DNA treads41. In the case of EFNB3, in silico search shows that it has a highly ranked enhancer at 27.9 kb 5′ of its start site43. Therefore, it is entirely possible that there is another regulatory element in the LD block which spans from 48.5 to 37.9 kb 5′ of the EFNB3 start site, despite that there are two other genes (TP53 and WRAP53) between the putative regulatory element and EFNB3.
To prove experimentally that EFNB3 mutations in the hypogonadic patients could indeed cause hypertension, we investigated the BP in EFNB3 KO mice. The male EFNB3 KO mice are normotensive26. The BP of the castrated WT males was decreased compared to uncastrated WT ones26. This is consistent with other reports that reduced testosterone level is often associated with lower BP in animal models44. However, in KO males, castration had no such BP-lowering effect, and as a result, the BP of the castrated KO mice was significantly higher than that of the castrated WT mice26. This in vivo result in mice suggests that male patients with EFNB3 loss-of-function mutations could not enjoy the BP-lowering benefit of reduced testosterone levels, which could be due to either ageing or other medical conditions. For these patients, if they suffer from other detrimental genetic or environmental influences that cause BP increase, the lost protection will render them hypertensive, as hypertension is a polygenic and multifactorial disease. As a consequence, compared to individuals without such EFNB3 loss-of-function mutations, these patients will have relatively increased hypertension risks. This is consistent with the results of our current human genetic study, in which we reveal that three EFNB3 SNPs are significantly associated with hypertension in hypogonadic patients.
Similar studies in patient cohorts from different ethnic backgrounds are warranted to generalize the conclusion that loss-of-function mutations in the EPHB6 signalling pathway molecules is a relative hypertension risk, and that such risk will only be materialized after testosterone levels are reduced.
References
Eph Nomenclature Committee. Unified nomenclature for Eph family receptors and their ligands, the ephrins. Cell 90, 403–404 (1997).
Pasquale, E. B. Eph-ephrin bidirectional signaling in physiology and disease. Cell 133, 38–52 (2008).
Flanagan, J. G. & Vanderhaeghen, P. The ephrins and Eph receptors in neural development. Annu Rev Neurosci 21, 309–345 (1998).
Luo, H., Wan, X., Wu, Y. & Wu, J. Cross-linking of EphB6 resulting in signal transduction and apoptosis in Jurkat cells. J. Immunol 167, 1362–1370 (2001).
Luo, H., Yu, G., Wu, Y. & Wu, J. EphB6 crosslinking results in costimulation of T cells. J. Clin Invest 110, 1141–1150 (2002).
Yu, G., Luo, H., Wu, Y. & Wu, J. Ephrin B2 induces T cell costimulation. J. Immunol 171, 106–114 (2003).
Yu, G., Luo, H., Wu, Y. & Wu, J. Mouse ephrinB3 augments T-cell signaling and responses to T-cell receptor ligation. J. Biol Chem 278, 47209–47216 (2003).
Luo, H., Yu, G., Tremblay, J. & Wu, J. EphB6-null mutation results in compromised T cell function. J. Clin Invest 114, 1762–1773 (2004).
Yu, G., Luo, H., Wu, Y. & Wu, J. EphrinB1 is essential in T-cell-T-cell co-operation during T-cell activation. J. Biol Chem 279, 55531–55539 (2004).
Wu, J. & Luo, H. Recent advances on T-cell regulation by receptor tyrosine kinases. Curr Opin Hematol 12, 292–297 (2005).
Yu, G., Mao, J., Wu, Y., Luo, H. & Wu, J. Ephrin-B1 is critical in T-cell development. J. Biol Chem 281, 10222–10229 (2006).
Luo, H. et al. Efnb1 and Efnb2 proteins regulate thymocyte development, peripheral T cell differentiation, and antiviral immune responses and are essential for interleukin-6 (IL-6) signaling. J. Biol Chem 286, 41135–41152 (2011).
Luo, H. et al. Ephrinb1 and Ephrinb2 are associated with interleukin-7 receptor α and retard its internalization from the cell surface. J. Biol Chem 286, 44976–44987 (2011).
Jin, W., Luo, H. & Wu, J. Effect of reduced EPHB4 expression in thymic epithelial cells on thymocyte development and peripheral T cell function. Mol Immunol 58, 1–9 (2014).
Hu, Y. et al. Role of EFNB1 and EFNB2 in Mouse Collagen-Induced Arthritis and Human Rheumatoid Arthritis. Arthritis Rheumatol 67, 1778–1788 (2015).
Luo, H. et al. EphrinB1 and EphrinB2 regulate T cell chemotaxis and migration in experimental autoimmune encephalomyelitis and multiple sclerosis. Neurobiol Dis 91, 292–306 (2016).
Batlle, E. et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell 111, 251–263 (2002).
Davy, A., Bush, J. O. & Soriano, P. Inhibition of gap junction communication at ectopic Eph/ephrin boundaries underlies craniofrontonasal syndrome. PLoS Biol 4, e315, https://doi.org/10.1371/journal.pbio.0040315 (2006).
Zhao, C. et al. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab 4, 111–121 (2006).
Wang, H. U., Chen, Z. F. & Anderson, D. J. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93, 741–753 (1998).
Konstantinova, I. et al. EphA-Ephrin-A-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell 129, 359–370 (2007).
Hashimoto, T. et al. Ephrin-B1 localizes at the slit diaphragm of the glomerular podocyte. Kidney Int 72, 954–964 (2007).
Dravis, C. et al. EphB2 and ephrin-B2 regulate the ionic homeostasis of vestibular endolymph. Hear Res 223, 93–104 (2007).
Luo, H. et al. Receptor tyrosine kinase Ephb6 regulates vascular smooth muscle contractility and modulates blood pressure in concert with sex hormones. J. Biol Chem 287, 6819–6829 (2012).
Wu, Z. et al. Possible role of Efnb1 protein, a ligand of Eph receptor tyrosine kinases, in modulating blood pressure. J. Biol Chem 287, 15557–15569 (2012).
Wang, Y. et al. Estrogen and testosterone in concert with EFNB3 regulate vascular smooth muscle cell contractility and blood pressure. Am J Physiol Heart Circ Physiol 310, H861–H872 (2016).
Wang, Y. et al. EPHB4 protein expression in vascular smooth muscle cells regulates their contractility, and EPHB4 deletion leads to hypotension in mice. J. Biol Chem 290, 14235–14244 (2015).
Wang, Y. et al. Reduced blood pressure after smooth muscle EFNB2 deletion and the potential association of EFNB2 mutation with human hypertension risk. Eur J Hum Genet 24, 1817–1825 (2016).
Tremblay, J. et al. Evidence from single nucleotide polymorphism analyses of ADVANCE study demonstrates EFNB3 as a hypertension risk gene. Sci Rep 7, 44114, https://doi.org/10.1038/srep44114 (2017).
Wang, Y. et al. The role of GRIP1 and ephrin B3 in blood pressure control and vascular smooth muscle cell contractility. Sci Rep 6, 38976, https://doi.org/10.1038/srep38976 (2016).
Traish, A. M., Miner, M. M., Morgentaler, A. & Zitzmann, M. Testosterone deficiency. Am J Med 124, 578–587 (2011).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res 12, 996–1006 (2002).
de Bakker, P. I. et al. Efficiency and power in genetic association studies. Nat Genet 37, 1217–1223 (2005).
Howie, B., Marchini, J. & Stephens, M. Genotype imputation with thousands of genomes. G3 (Bethesda) 1, 457–470 (2011).
The 1000 Genomes Consortium et al. A global reference for human genetic variation. Nature 526, 68-74 (2015).
Purcell, S. et al. PLINK: a toolset for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575 (2007).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Jul 16, 2013
The Broad Institute of MIT and Harvard. GREx Portal, https://www.gtexportal.org/home/ (2017)
Forrester, W. C. et al. A deletion of the human beta-globin locus activation region causes a major alteration in chromatin structure and replication across the entire beta-globin locus. Genes Dev 4, 1637–1649 (1990).
Antes, T. J. et al. Identification and characterization of a 315-base pair enhancer, located more than 55 kilobases 5′ of the apolipoprotein B gene, that confers expression in the intestine. J. Biol Chem 275, 26637–26648 (2000).
Spitz, F. Gene regulation at a distance: From remote enhancers to 3D regulatory ensembles. Semin Cell Dev Biol 57, 57–67 (2016).
Sanyal, A., Lajoie, B. R., Jain, G. & Dekker, J. The long-range interaction landscape of gene promoters. Nature 489, 109–113 (2012).
Weizmann Institute of Science. GeneCards, Human gene database, http://www.genecards.org/cgi-bin/carddisp.pl?gene=EFNB3 (2018)
Ely, D. L., Salisbury, R., Hadi, D., Turner, M. & Johnson, M. L. Androgen receptor and the testes influence hypertension in a hybrid rat model. Hypertension. 17(6 Pt 2), 1104–10 (1991).
Acknowledgements
This work was supported by the Fonds de recherche du Québec–Santé, the Natural Sciences and Engineering Research Council of Canada (RGPIN-2017-04790), the Canadian Institutes of Health Research (MOP 123389), and the J.-Louis Lévesque Foundation to J.W. This study was also made possible by a group grant from the National Sciences Foundation of China (#81361120264) to J.S., S.H., T.W. and J.W., and a grant from the National Sciences Foundation of China to Y.W. (#81700363). The funders provided support in the form of salaries and experimental costs, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the “author contributions”.
Author information
Authors and Affiliations
Contributions
T.W., H.L., W.E.B., J.W., J.S. and S.H. generated the concept and initiated this project. T.W., B.Z., Y.Y., H.W., Z.X., Y.W., J.S. and S.H. conducted experiments. J.R., F.M. and M.R.T. performed genetic data analysis. T.W., J.R. and J.W. drafted the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, T., Zhang, BQ., Raelson, J. et al. Analysis of the association of EPHB6, EFNB1 and EFNB3 variants with hypertension risks in males with hypogonadism. Sci Rep 8, 14497 (2018). https://doi.org/10.1038/s41598-018-32836-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32836-x
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
