
Abstract
Tinospora crispa is a popular traditional herbal plant commonly used throughout the world for treatment of various diseases, in particular type 2 diabetes mellitus. We report here a new case of toxic hepatitis in a 57-year old male patient in the French West Indies following the consumption of two aqueous extracts of fresh Tinospora crispa stems. It thus differs from two previously reported cases that concerned the chronic intake of powdered dry stems delivered in solid oral dosage forms (i.e. pellets and tablets). Liquid Chromatography-Diode Array Detection-Mass Spectrometry (LC/DAD/MS) analyses were performed on an aqueous extract of the offending sample that mimics the swallowed preparation. They revealed the presence of species-specific molecular marker borapetoside C (1) and thus enabled an unambiguous phytochemical identification. The exploration of tandem MS/MS data obtained by ultra-high performance liquid chromatography/electrospray ionization quadrupole time-of-flight mass spectrometry (UHPLC-ESI-QTOF-HRMS) allowed the identification of 17 additional cis-clerodane-type furanoditerpenoid lactones, analogues of 1. These results support the hypothesis that the mechanisms underlying hepatotoxicity of Tinospora crispa are the same as those encountered with furanoditerpenoids-containing plants such as Teucrium chamaedrys or Dioscorea bulbifera. In the context of type 2 diabetes treatment, we recommend that Tinospora crispa intake should be more closely monitored for signs of hepatotoxicity.
Similar content being viewed by others


Introduction
Tinospora crispa (L.) Hook. f. & Thomson belongs to the botanical family of Menispermaceae. The plant is widely distributed in primary rain forests in Southeast Asia, China and Africa. It is also present in French Guyana and the French West Indies as an ornamental plant for gardens. It is a popular traditional medicinal plant in Asia, mainly as a stem decoction1, for the treatment of diabetes, jaundice, rheumatism, urinary disorders, fever, malaria or hypertension. Its potential for the treatment of diabetes is of interest for the following reasons: the growing number of people with type 2 diabetes in the world, the cost of conventional glucose-lowering medications (and the limitations related to their efficacy on glycaemic control in the long term/run), and the high consumer demand for alternative therapy. Promising results have been recently observed both in vitro and in vivo by several teams of researchers, raising hope for future application in diabetology2. However, conflicting results have been obtained in clinical studies and its safety profile remains to be determined. Indeed, a potential hepatoxicity has been raised by two case reports following the chronic consumption of pellets or tablets containing T. crispa in Asia3,4. Here we report the new case of a patient having developed toxic hepatitis due to the consumption of two aqueous extracts of T. crispa stem in the French West Indies. We discuss the benefit/Risk of the use of this plant as an alternative therapy for the treatment of diabetes in the light of current literature. Liquid Chromatography-Diode Array Detection-Mass Spectrometry (LC/DAD/MS) and Liquid Chromatography-tandem Mass Spectrometry (LC/MS/MS) profiling enabled an unambiguous phytochemical identification of the incriminated sample and revealed the presence of putative toxins, namely the cis-clerodane-type furanoditerpenoid lactones in an aqueous stem extract similar to the traditional preparation.
An expeditious targeted qualitative analysis of these secondary metabolites was performed by combining classical LC/MS/MS data exploration (i.e. tentative identification of the compounds based on the exact mass and fragmentation pathways/formal identification by comparison of the retention time (tR), UltraViolet (UV) spectrum and tandem mass spectrometry (MS/MS) data with a reference compound) and molecular networking analysis (MS2-MN)5,6. This has allowed to observe possible similarities between compound spectra and thus to study shared fragment ions.
Case Report
A 57-year-old male patient, with no personal or family history of liver disease or alcohol addiction, was admitted for acute hepatitis with cholestasis and cytolysis. Three weeks before, he had started consuming an aqueous extract of “lian-sepent”, a traditional medicine corresponding to T. crispa according to local ethnobotanists. This herbal remedy was supposed to detoxify his liver. It consisted of a piece of T. crispa stem put into a bottle of water and drunk regularly over the next two days. Two weeks later, the patient prepared a similar aqueous extract of T. crispa and consumed it over two days. Following his last intake, the patient felt fever and asthenia for one week and went to the emergency room after occurrence of dark urine. On admission, he suffered from jaundice, was not overweight, and had normal vital signs. Biological tests revealed evidence of hepatocellular damage (ALT 1923 U/L; AST 873 U/L) and cholestasis (γGT 155 U/L). Abdominal ultrasonography was normal with no hepatomegaly or lithiasis. The results of laboratory testing disclosed no serological arguments for viral hepatitis (hepatitis A virus, hepatitis B virus, hepatitis C virus, cytomegalovirus, Epstein-Barr virus and varicella zoster virus). The patient was discharged two weeks after admission and evolution was marked by the regression of jaundice and progressive decrease in liver function tests without any specific treatment (Table 1).
Laboratory Investigations
Identification of plant sample
The sample provided by the patient consisted of fresh stems of T. crispa as ascertained by comparison of both macroscopic botanical features and phytochemical profile, with those of an authentic herbal reference standard4. As shown in Fig. 1, stems display a prominent tuberculate surface absent in the widespread related medicinal species T. cordifolia (Willd.) Miers and T. sinensis (Lour.) Merr. (synonyme of T. malabarica (Lam.) Hook f. & Thomson)7.

Sample of Tinospora crispa involved in toxic hepatitis (Author: X.C.).
Chromatographic profiles of both the analysed and the reference samples were next established by High-Performance Liquid Chromatography with Diode-Array Detection-Mass Spectrometry HPLC-DAD-MS for their corresponding CH2Cl2 extracts (Fig. 2).

Reverse Phase-HPLC-UV chromatograms of CH2Cl2 extracts: (lower trace: 1): offending sample, (upper trace: 3): reference sample; of aqueous extract: (middle trace: 2); with an offset of 3 min. Embedded data (from right to left): borapetoside C (1): chemical structure, UV/Visible spectrum and positive Electrospray Ionisation-Mass Spectrometry (ESI-MS) spectrum.
Borapetoside C (1)8,9 (synonym tinocrisposide), a species-specific molecular marker of Tinospora crispa was unambiguously identified in the herbal reference standard as well as in the suspect sample by comparison of the tR, UV spectrum and MS data with a reference compound isolated from the plant (tR 55.2 min; λmax: 214 nm; m/z 559: Na adduct [M + Na]+, 537: pseudo-molecular ion [M + H]+; fragment ions at m/z 375, 357, 339, 325, 311, 307, 297, 279, 251, 205, 187 and 159). Interestingly, this compound appeared as the main peak on the LC-UV (214 nm) chromatogram of the analysed sample (Fig. 2, lower chromatogram). The amount of this compound was clearly found higher in the fresh stems than in the older reference sample, moreover consisting of dried stems (Fig. 2, upper chromatogram).
Phytochemical analysis of an aqueous extract that mimics the traditional preparation
We next investigated whether borapetoside C (1) and other potentially toxic related clerodane furanoditerpenoids could be identified in an aqueous extract of fresh stems that mimics the traditional preparation.
First of all, the peak assigned to 1 was also found predominant in the chromatogram obtained by HPLC/DAD/MS and depicted in Fig. 2 (middle chromatogram). However, this finding was not really surprising since glycosides are essentially water-soluble.
Subsequently, a targeted qualitative analysis of the furanoditerpenoids structurally related to 1 was carried out in two ways: by using ultra-high performance liquid chromatography/electrospray ionization quadrupole time-of-flight mass spectrometry (UHPLC-ESI-QTOF-MS/MS) and by analysing the generated data on the basis of the exact mass of the compounds and their fragmentation pathways, in combination with a molecular networking approach for the whole dataset processing and especially their filtering. For this purpose, the data analysis portal of the Global Natural Products Social Molecular Networking (GNPS) web-based platform (http://gnps.ucsd.edu) was used. This approach constitutes a powerful and innovative tool for the dereplication of natural products and consequently for the identification of new ones. Building of molecular networks is based on relatedness analysis within tandem MS/MS data. It enables to visualize them as a molecular network clustering together data for structurally related molecules. MS/MS spectra are represented as nodes connected together by edges symbolizing close similarities between them5,6. Consequently, introducing this step in the workflow allowed an efficient data filtering and facilitated the targeting of compounds structurally analogous to borapetoside C (1).
The chromatographic profile of the aqueous extract obtained by ultra-high performance liquid chromatography/electrospray ionization quadrupole time-of-flight mass spectrometry (UHPLC-ESI-QTOF-HRMS) was found similar to the one previously obtained by HPLC/DAD/MS (see Fig. 2). A representative base peak chromatogram (BPC) with numbered peaks (No. 1–18), corresponding to the furanoditerpenoids detected in this study, is illustrated in Fig. 3. In these conditions, the peak (No. 15) corresponding to borapetoside C (1) was detected with a tR of 13.7 min (data matching those observed for standard). Major fragment ions observed in the MS/MS spectra of 1 are listed in Table 2, and the logical fragmentation pathways for this compound are described in Fig. 4.

UHPLC-ESI-QTOF-HRMS base peak chromatogram of the T. crispa stems aqueous extract (numbered peaks corresponds to the furanoditerpenoids identified in this study).

Proposed fragmentation pathways of borapetoside C (1).
The molecular network generated with the MS/MS data acquired from both the CH2Cl2 and aqueous extracts is depicted in Fig. 5. Within the molecular network we focused our attention on the cluster exhibiting a central node associated with the precursor mass and the tR of borapetoside C (1). It should be noted that under the adopted conditions of “one-shot” analysis (i.e. without use of enriched fractions and optimization of the collision energy) several nodes were found to correspond exclusively to fragment ion spectra and are thus labelled with the corresponding precursor mass. For instance, nodes labelled with the m/z values 375.180 and 357.170 correspond to fragment ion spectra of borapetoside C (1). However, to a single node may correspond several compounds either isomeric (e.g. m/z 685.270) or sharing a common fragment ion (e.g. m/z 553.228), then differentiated by their specific retention times. Finally, scrutinizing the cluster, in combination with complete inspection of MS/MS spectra and extracted ions chromatograms supported the presence of 17 additional furanoditerpenoid analogues of 1, corresponding to peaks notified as No 1-14 and 16-18 in the base peak chromatogram (Fig. 3). Major fragment ions of these compounds are also listed in Table 2, as well as the parent ions values observed in the molecular network (bolded values).

Molecular network generated with UHPLC-ESI-QTOF-HRMS2 data from the CH2Cl2 and aqueous extracts of T. crispa stems and visualized using Cytoscape software. Cosine similarity score cutoff was of 0.6. Nodes are labelled with parent or precursor mass value and their size are linked to the number of spectra (molecular formula of corresponding ions are indicated beside nodes). Nodes coloring: nodes are represented as pies and each color represents a group of spectrum files associated with an extract: red: G1/aqueous extract; blue: G2/CH2Cl2 extract. Edges are annotated with mass difference and their thickness depends on the cosine score ranging between 0.6 (minimum accepted similarity between spectra) and 1 (maximum similarity between spectra). See Material and Methods for supplementary details.
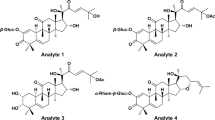
Tentative identification of furanoditerpenoids analogues of 1 was based on the determination of molecular formulas deduced from accurate m/z values and fragmentation patterns and additionally through dereplication using database search including the Dictionary of Natural Products, Reaxys and SciFinder. Chemical structures of the putatively identified furanoditerpenoids are presented in Fig. 6.

Chemical structures of furanoditerpenoids identified in T. crispa stem aqueous extract.
Borapetoside C (1) showed a [M + H]+ ion at m/z 537.2331 (calculated for C27H37O11: 537.2330, Δppm = 0.3) and fragment ions at m/z 375.1802 and 357.1699, consistent with the loss of the 2-deoxyglucosyl (162 uma) and β-D-glucosyl (180 uma) moieties, respectively. Fragment ions produced by the successive elimination of methanol (32 uma) and carbon monoxide (28 uma) were characteristic of the presence of a carbomethoxy group (60 uma). For example, the fragment ions observed at m/z 325.1433 and 297.1483 may be formed from the protonated ion at m/z 357.1699 as well as the fragment ion at m/z 251.1431 may be produced from the cationic ion at m/z 311.1642. The concomitant neutral loss of CO2 and C6H6O (attributed to 3-vinylfuran), accounting for 138 uma, was typical fragmentation pattern of the 6-furyl-tetrahydro-2H-pyran-2-one structure and may explain the formation of fragment ions observed at m/z 205.0859, 187.0754 and 159.0803.
Compounds eluting at 11.8, 13.0, 13.1, 13.4, 13.5, 14.1, 15.6 and 16.7 min (peaks 9, 11–14, 16–18, respectively) and borapetoside C (1) shared the same fragmentation patterns and should consequently display a close similarity in their structures.
Compounds corresponding to peaks 13 and 14 exhibited the same pseudomolecular ion at m/z 537.2332/537.2338, respectively (calculated for C27H37O11: 537.2330, Δppm = 0.3/1.4) as that of 1 and both could be tentatively assigned to borapetoside E10,11 or to unknown stereoisomers of borapetosides C and E. Peaks 9, 11 and 12, could be tentatively assigned to borapetoside D (=6′ → 1″)-O-β-D-glucopyranosyl borapetoside E)10,11 or to unknown isomers of this molecule including O-hexosyl borapetoside C derivatives. They exhibited a same protonated ion at m/z 699.2842/699.2859/699.2866, respectively (calculated for C33H47O16: 699.2859, Δppm = −2.4/0.0/1.0). Peaks No 17 (tR of 15.6 min) and 18 (tR of 16.7 min) may correspond to crispenes A and D12, respectively. These compounds showed a [M + H]+ ion at m/z 343.1544 (calculated for C20H23O5: 343.1540, Δppm = 1.1) and 375.1792 (calculated for C21H27O6: 375.1802, Δppm = −2.8), respectively. Finally, the compound corresponding to peak No 16 (tR of 14.1 min) displayed a protonated ion at m/z 681.2749 (calculated for C33H45O15: 681.2753, Δppm = −0.6) and has not been previously reported. The mass difference observed between the protonated ions of this compound and that of 1 was 145 uma, which could be reasonably attributed to one additional ester group of a hydroxylated dicarboxylic acid (C6H10O5).
A second group of borapetoside C analogues is represented by compounds bearing a hydroxyl group at the C-2 position such as borapetoside B. They all shared common fragmentation pathways with probably similar reactions to those described for 1, as illustrated in Fig. 7 for borapetoside B. However, protonated parent ions and fragment ions displayed a characteristic difference of mass of 16 uma attributed to oxygen in comparison to analogous ions observed in the tandem mass spectra of 1. Compounds belonging to this group are significantly more hydrophilic and were eluted between 7.0 and 12.0 min (peaks 1–6 and 10).

Proposed fragmentation pathways of borapetoside B (1).
Compounds corresponding to peaks 5 (tR of 9.1 min) and 6 (tR of 9.3 min) exhibited the same pseudomolecular ion at m/z 553.2277/553.2279, respectively (calculated for C27H37O12: 553.2280, Δppm = −0.5/−0.1) and both could be tentatively assigned to borapetoside B8 or its reported isomers (i.e. rumphioside I11 and the diastereosiomer 313). Peak No 1 (tR of 7.0 min) may correspond to borapetoside H2 since it displayed a protonated parent ion at m/z 715.2828 (calculated for C33H47O17: 715.2810, Δppm = 2.8). Peaks No 3 (tR of 8.2 min) and 4 (tR of 8.7 min) displayed a same [M + H]+ ion at m/z 685.2687/685.2700, respectively (calculated for C32H45O16: 685.2702, Δppm = −2.2/−0.3) and both may be tentatively assigned as the reported (2R,5R,6R,8S,9S,10S,12S)-15,16-epoxy-2-hydroxy-6-O-{β-D-glucopyranosyl-(1 → 6)-α-D-xylopyranosyl}-cleroda-3,13(16),14-trien-17,12-olid-18-oic acid methyl ester (2)13 or an unknown isomer of this compound. Peak No 10 (tR of 12.0 min) could be tentatively identified as borapetol B10, the aglycon moiety of borapetoside B. This compound exhibited a [M + Na]+ at m/z 413.1570 (calculated for C21H26NaO7: 413.1571, Δppm = −0.2). Compound eluted at tR = 7.9 min (peak No 2) showed a [M + Na]+ at m/z 593.2209 (calculated for C27H38NaO13: 593.2205, Δppm = −1.2). This new compound is most likely the corresponding analogue of either borapetoside B or its isomers, with the lactone ring opened and converted to 5-hydroxypentanoic acid moiety.
A last group corresponds to compounds eluted at 9.7 (peak No 7) and 11.7 min (peak No 8). These compounds shared some fragment ions with borapetoside C (1) and its close structural analogues particularly those at m/z 251, 159 and 131, but also exhibited in their MS/MS tandem spectra several specific fragment ions. Peak No 8 was tentatively assigned to the non-glycosidic furanoditerpenoid crispene B12, characterized by the presence of a carboxyl group at C-4 in place of a carbomethoxyl group. This compound showed a [M + Na]+ at m/z 383.1465 (calculated for C20H24NaO6: 383.1465, Δppm = −0.1) and a characteristic protonated fragment ion at m/z 343.1543 which corresponds to the loss of H2O ([M-H2O+H]+). Compound eluting at 9.7 min exhibited quasimolecular molecular ions at m/z 545.1998 ([M + Na]+: calculated for C26H34NaO11: 545.1993, Δppm = + 0.9) and at m/z 567.1804, which corresponds to a deprotonated carboxyl group ([M-H + 2Na]+: calculated for C26H33Na2O11: 567.1813, Δppm = −1.5), as well as fragment ions at m/z 383.1468 ([M-162 + Na]+), 365.1358 ([M-180 + Na]+) and 343.1541 [M-180 + H]+. We concluded from these data that this compound was an unknown O-glycosyl derivative of crispene B or of its isomers (such as, for example, the corresponding carboxylic acid of 1).
Interestingly, apart from mass and chromatographic information provided by molecular network exploration, node coloring indicates that all these compounds were detected in the aqueous extract. This observation is consistent with the presence of highly polar/hydrophilic molecules such as, for instance, borapetosides D and H which bear two sugar units, as well as borapetoside B and rumphioside I with a free hydroxyl group at the C2 position.
Discussion
Among the large spectrum of traditional use of T. crispa reported in the literature, its anti-diabetic activity has raised a special interest for researchers all around the world considering the growing number of type 2 diabetes cases, all the more so as its use in South-Asia has been widespread for a long time. We have discovered that the plant is also consumed in the French West Indies (and probably throughout the Caribbean Arc due to the historic intermingling of Caribbean populations). Indeed, a survey has shown that 60% of diabetic patients in Martinique (French West Indies) use plants in addition to their glucose lowering drugs, among them T. crispa was the second plant the most used14. However, like several alternative medicine therapies for diabetes, finding reliable information about safety and benefits remains difficult. Notably, adverse effects associated with traditional medicine are often not well documented.
In this report, we present a case of toxic hepatitis following occasional consumption of a stem aqueous extract of the T. crispa. Hepatitis induced by T. crispa is established, based on absence of medical history, clinical exam and biological results. We noted that the patient had been exposed to several pesticides the next day after the last ingestion of the aqueous extract. Yet, to the best of our knowledge acute cutaneous or respiratory exposure to those pesticides (containing myclobutanil, glyphosate, acetamipride, abamectin and linuron) could not explain the onset of hepatitis. Hepatitis was reversible and biological hepatic enzymes returned to normal after a few weeks without any specific treatment. This conclusion was corroborated by calculation of Roussel Uclaf Causality Assessment Method (RUCAM) score. With a calculated score of +6, the causal relationship between T.c. aqueous extract intake and hepatotoxicity was “probable”15.
Two previous cases of acute hepatitis have been reported with T. crispa but with a different method of administration consisting of chronic use of tablets or pellets of the plant. The first case of hepatitis concerned a 37-year-old woman who had consumed T. crispa tablets (bought in Indonesia) for 10 weeks3. The second case concerned a 49-year-old man who had orally taken pellets of T.crispa (bought on a Vietnamese market4) for 4 weeks. In both cases, hepatitis was reversible after a few weeks. These case reports are in accordance with changes in biochemical parameters observed during some clinical trials assessing the effect of T. crispa in diabetic patients and in patients with metabolic syndrome. Marked elevation of liver enzymes (that returned to normal after discontinuing T. crispa) has been observed in 2 of the 20 patients treated for 6 months with a capsule form at a dosage of 1 gram thrice daily16. Similarly, a double-blind, placebo-controlled trial using a crossover design found an elevation of more than 3 times baseline levels of ALT and AST in 6 of the 36 patients who received 250 mg T. crispa dry powder capsule twice a day for 2 months17.
We hypothesize that two mechanisms may have contributed to hepatotoxicity induced by T. crispa, considering the close structural similarity observed between borapetosides and i. furanoditerpenoids like 8-Epidiosbulbin E acetate (EEA) and ii. teucrin A or teuchmaedryn A. The first mechanism is direct liver toxicity induced by metabolic activation of T. crispa furanoditerpenoids. Such a mechanism of toxicity has been described with EEA, a norclerodane furanoditerpenoid structurally very similar to borapetosides18. Cytochrome P450 metabolic activation of the furan moiety of EEA may be responsible for the formation of electrophilic species, leading to a dose-dependent hepatotoxicity. Other similar examples of bioactivation of furanoditerpenoids correlated with hepatotoxicity are reported in literature18,19. The second toxicological mechanism may be idiosyncratic, as it has been reported that only a few people exposed may develop hepatitis16,17. Such a mechanism may be suspected in our case, as hepatitis occurred after reexposure to T. crispa and was associated with fever, a typical symptom of idiosyncratic immunoallergic toxic hepatitis. Moreover, there is a close structural analogy between borapetosides and teucrin A or teuchmaedryn A, two clerodanes found in Teucrium chamaedrys L. (Lamiaceae). In-vivo and in-vitro studies have demonstrated that those teucrin A or teuchmaedryn A induce hepatotoxicity, consecutive to both direct toxicity and a secondary immune reaction with antoantibody formation20. Future studies using isolated compounds will be necessary to ascertain the exact mechanism of toxicity of the furanoditerpenoids from Tinospora crispa stems.
The present UHPLC-ESI-QTOF-MS/MS metabolomic investigation performed on the aqueous stem extract of T. crispa led to the detection of 18 furanoditerpenoids structurally related to the major compound, namely borapetoside C (1). A targeted analysis of MS/MS data was facilitated by introducing a molecular networking approach in the data analysis workflow. MN is becoming one of the most efficient tools for the analysis of untargeted MS/MS data, allowing dereplication of complex extracts and exploration of molecular diversity21. We have showed here that MN is also valuable for targeted metabolite fingerprinting. To the best of our knowledge this is the first example of application of MN for investigating plant suspected of being toxic and toxins thereof.
Hepatic trouble should be put in perspective with the potential hypoglycaemic effect of the plant. Promising results have been obtained by in vitro and in vivo tests with T. crispa. Noor et al. have observed insulin secretory rates in alloxan-diabetic rats and insulin release from rat islets and HIT-T15 beta cells in vitro with the extract of stem plant22. Since this publication, its hypoglycaemic mechanism of action has been attributed to diterpenoids (borapetol B, borapetoside A and C) isolated from the plant that could stimulate insulin release from pancreatic β-cell. Using an oral glucose tolerance test, an increase of 2-fold of plasma insulin release has been observed after oral administration of borapetol B to Wistar rats and spontaneously type 2 diabetic Goto-Kakizaki rats compared to placebo23. Similarly, administration of borapetoside A to diet-induced type 2 diabetes mellitus mice lowered the plasma glucose level in a dose-depended manner with results similar to the administration of metformin for the dose range between 0.3 and 10 mg/kg24. A potential mechanism of improvement of peripheral glucose uptake, notably via an increase in glucose utilization of skeletal muscle and liver, has also been advanced following in vitro tests24,25,26. To date, these promising non-clinical studies have unfortunately not been confirmed by clinical trials applied especially to type 2 diabetic patients. Indeed, Klangjareonchai et al. showed that glucose and insulin areas under the curve were not different with or without ingestion of 125 or 250 g of T. crispa dry power capsule27, while Sangsuwan et al. in a randomized, double blind, placebo-controlled trial found no difference on HbA1c or fasting plasma glucose between patients treated with 1 gram thrice daily Tinospora crispa powder in capsule for 6 months and patients treated with placebo16.
In total, the Benefit-Risk of the use of Tinospora crispa for the treatment of type 2 diabetes appears negative at this step, despite the hope raised by non-clinical studies. There remain uncertainties on the efficacy on blood glucose control in real-life setting while the risk of hepatic trouble is established, both for an acute and for a chronic use and independently of the method of administration (stem in powder capsule or used as an aqueous extract). Furthermore, toxic mechanisms may associate both dose-response relationship and idiosyncratic effects, therefore making therapeutic use of T. crispa hazardous, in so far as diabetic patients are often treated simultaneously with statins, drugs known to potentially increase liver enzymes.
Conclusion
Chronic or occasional use of T. crispa stem could induce toxic hepatitis, reversible after a few weeks without any specific treatment. Despite its promising results suggesting an increase in insulin release in non-clinical studies, its traditional use should be avoided for diabetic patients for lack of demonstrated benefit obtained by dedicated well-controlled clinical trials. Several furanoditerpenoids have been detected and putatively identified by UHPLC-ESI-QTOF-MS/MS in an aqueous extract of fresh stems that mimics the traditional preparation by combining classical data exploration and molecular networking approach. Even in non-optimized conditions of data acquisition, molecular networking constitutes a powerful and useful tool facilitating the data filtering. Further studies are still needed to confirm the putative toxicity of furanoditerpenoids and to elucidate subjacent mechanisms.
Materials and Methods
Questioning and biochemical analyses
Informed consent was obtained from the patient. The Faculté de Pharmacie de Paris, Université Paris Descartes approved the experimental protocol and it was carried out in accordance with all relevant guidelines and regulations. Biochemical analyses were performed by standard methods using automated techniques.
Standards
Authentic herbal reference standard
Stems were originally collected in 2013 in the Botanical Garden of the Hanoi University of Pharmacy in Hanoi, Vietnam and authenticated par Plant Records Officer Dr Quoc Huy Nguyen. A voucher specimen is stored in the herbarium collection of the François Tillequin Museum of Materia Medica (Faculty of Pharmacy, Paris) under the N°02486. Borapetoside C. Borapetoside C was isolated from T crispa in our laboratory by column chromatography and unambiguously identified by 1H, 13NMR and MS by comparison with literature data9,10.
Sample preparation
Dichloromethane extracts of T. crispa stem
Test and reference solutions were prepared in the same way by maceration of stems in methylene chloride for 72 h, followed by filtration, then drying under vacuum (Drug Extract Ratio (DER): about 47:1 and 24:1, respectively). Aqueous extracts of T. crispa stem. Given the limited amount of material available (i.e. 1.2 g) stems were boiled in water (10 mL) for 10 min and the resulting solution was filtered prior to lyophilisation (DER: about 100:1). Moreover, the complete solubility of the lyophylisate in deionized water at ambient temperature (20 °C) was checked at a concentration of 5.6 mg/mL.
Chromatographic conditions
RP-HPLC-UV (ESI-MS) conditions
HPLC-System: Thermo Scientific Dionex Ultimate 3000®, Courtaboeuf, France) with binary pump, PDA detector, autosampler and Chromeleon software. Detection: 214 nm. Injection volume: 20 μL. Column: Zorbax SB®-C18 (5 μm, 4.6 × 250 mm), equipped with a pre-column and thermostated at 25 °C. Mobile Phase: Gradient: [MeOH:Water-0.05% TFA]: 10:90 → 80:20 within 60 min). Flow: 0.6 mL/min. All solutions were prepared in MeOH (2 mg/mL) and filtered through 0.2 μm nylon filter disk prior the injection. ESI-MS spectrum (Thermo-Scientific Surveyor MSQ Plus single-quadrupole mass detector (Thermo Fisher Scientific, Courtaboeuf, France)).
UHPLC-HRMS and MS2 experiments
Separations were performed using an Ultimate 3000 RSLC system equipped with a binary pump, an autosampler and a thermostated column compartment, equipped with a diode array detector (195–800 nm) (Dionex, Germering, Germany). Components were separated on a C18 Luna Omega column of 150 mm × 2.1 mm with a particle size of 1.6 μm (Phenomenex, Le Pecq, France). The mobile phase was made up of 0.1% formic acid in water (phase A), and 0.08% formic acid in acetonitrile (phase B). A solvent gradient was applied as follows: 0–0.1 min: 3% B, 0.1–26 min: 3–80% B, 26–26.5 min: 80–95% B, 26.5–29.5 min: 95% B, 29.5–30 min: 95–3% B, and finally 30–33 min: 3% B. The column was introduced in a thermostated compartment heated at 40 °C. All solutions were prepared in MeOH (2 mg/mL) and filtered through 0.2 μm nylon filter disk prior the injection. The injection volume was set at 1 µL and the flow rate was set at 500 μL/min. MS experiments were carried out on a maXis UHR-Q-TOF mass spectrometer (Bruker, Bremen, Germany) in positive electrospray ionization (ESI) mode. Capillary voltage was set at +4.5 kV. The flows of nebulizing and drying gas (nitrogen) were respectively set at 2.0 bar and 9.0 L/min and drying gas was heated at 200 °C. Mass spectra were recorded in the range 50–1650 m/z. MS/MS experiments were conducted using data dependent acquisition (DDA) mode (auto-MS/MS) in a mass window from m/z 150–1200. Three precursor ions with intensities higher than 400 au were selected per fragmentation cycle among the most intense ions to be fragmented. These three precursor ions were allowed to be selected for two consecutive cycles and were then placed on an exclusion list for 0.05 min. The collision energy was set at 35 eV and was applied as follows: 88% of the collision energy was applied during half of the fragmentation cycle, and 117% of the collision energy was applied during the half remaining cycle time. Raw data were converted to mzXML format using CompassXport software (Bruker, Bremen, Germany) and then processed using MZmine version 228.
Molecular Network Analysis
A molecular network was created using the online workflow at GNPS (https://gnps.ucsd.edu/ProteoSAFe/static/gnps-splash.jsp)6. The data was filtered by removing all MS/MS peaks within +/−17 Da of the precursor m/z. MS/MS spectra were window filtered by choosing only the top 6 peaks in the +/−50 Da window throughout the spectrum. The data was then clustered with MS-Cluster with a parent mass tolerance of 0.05 Da and a MS/MS fragment ion tolerance of 0.05 Da to create consensus spectra29. Further, consensus spectra that contained less than 1 spectrum were discarded. A network was then created where edges were filtered to have a cosine score above 0.6 and more than 2 matched peaks. Further edges between two nodes were kept in the network if and only if each of the nodes appeared in each other’s respective top 20 most similar nodes. The spectra in the network were then searched against GNPS’ spectral libraries. The library spectra were filtered in the same manner as the input data. All matches kept between network spectra and library spectra were required to have a score above 0.7 and at least 6 matched peaks. Molecular network parameters are available at https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=2817c0f504f741f781f645eb3ee27d95.
Data Availability Statement
Data of LC-MS/MS analysis were deposited to MassIVE Public GNPS dataset (http://gnps.ucsd.edu/MSV000082664).
References
Ahmad, W., Jantan, I. & Bukhari, S. N. Tinospora crispa (L.) Hook. f. & Thomson: A Review of Its Ethnobotanical, Phytochemical, and Pharmacological Aspects. Front Pharmacol. 21, 7–59 (2016).
Thomas, A., Rajesh, E. K. & Kumar, D. S. The Significance of Tinospora crispa in Treatment of Diabetes Mellitus. Phytother Res. 30, 357–366 (2016).
Denis, G. et al. Prophylaxie antipaludéenne par plantes médicinales: hépatite toxique a Tinospora crispa. Thérapie. 62, 271–272 (2007).
Langrand, J. et al. Toxic hepatitis induced by a herbal medicine. Tinospora crispa. Phytomedicine. 21, 1120–1123 (2014).
Yang, J. Y. et al. Molecular Networking as a dereplication strategy. J. Nat. Prod. 76, 1686–1699 (2013).
Wang, M. et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 34, 828–837 (2016).
Ahmad, S. M., Hoot, S. B., Qazi, P. H. & Verma, V. Phylogenetic patterns and genetic diversity of Indian Tinospora species based on chloroplast sequence data and cytochrome P450 polymorphisms. Plant Syst. Evol. 281, 87–96 (2009).
Fukuda, N., Yonemitsu, M. & Kimura, T. Isolation and structure elucidation of the five new furanoid diterpene glycosides borapetoside C-G. Liebigs Ann. Chem. 5, 491–495 (1993).
Lam, S.-H., Ruan, C. T., Hsieh, P.-H., Su, M.-J. & Lee, S.-S. Hypoglycemic diterpenoids from Tinospora crispa. J. Nat. Prod. 75, 153–159 (2012).
Fukuda, N., Yonemitsu, M. & Kimura, T. Studies on the constituents of the stems of Tinospora tuberculata BEUMEE. III. New diterpenoids, Borapetoside B and Borapetol B. Chem. Pharm. Bull. 34, 2868–2872 (1986).
Martin, T. S., Ohtani, K., Kasai, R. & Yamasaki, K. Furanoid diterpene glycosides from Tinospora rumphii. Phytochemistry. 42, 153–158 (1996).
Hossen, F., Ahasan, R., Haque, M. R., Begum, B. & Hasan, C. M. Crispene A, B, C and D, Four new clerodane type furanoid diterpenes from Tinospora crispa (L.). Pharmacogn. Mag. 12, S37–S41 (2016).
Choudhary, M. I. et al. Cis-Clerodane-type furanoditerpenoids from Tinospora crispa. J. Nat. Prod. 73, 541–547 (2010).
Longuefosse, J. L. Plantes médicinales martiniquaises à potentiel antidiabétique. http://www.academia.edu/5045689/Plantes_martiniquaises_%C3%A0_potentiel_antidiab%C3%A9tique (2004)
Danan, G. & Benichou, C. Causality assessment of adverse reactions to drugs-I. A novel method based on the conclusions of international consensus meetings: application to drug-induced liver injuries. J. Clin. Epidemiol. 46, 1323–1330 (1993).
Sangsuwan, C., Udompanthurak, S., Vannasaeng, S. & Thamlikitkul, V. Randomized controlled trial of Tinospora crispa for additional therapy in patients with type 2 diabetes mellitus. J. Med. Assoc. Thai. 87, 543–546 (2004).
Sriyapai, C., Dhumma-Upakorn, R., Sangwatanaroj, S., Kongkathip, N. & Krittiyanunt, S. Hypoglycemic effect of Tinospora crispa dry powder in outpatients with metabolic syndrome at King Chulalongkorn memorial hospital. J. Health Res. 23, 125–133 (2009).
Lin, D. et al. Role of metabolic activation in 8-epidiosbulbin E acetate-induced liver injury: mechanism of action of the hepatotoxic furanoid. Chem. Res. Toxicol. 29, 359–366 (2016).
Li, H., Peng, Y. & Zheng, J. Chapter Two – Activation and toxicities of furanoditerpenoids. Vol. 10. In: Fishbein, J. C., Heilman, J. (Eds), Advances in Molecular Toxicology. Academic Press, Cambridge, 55–97 (2016).
Gori, L. et al. Two Contemporary Cases of Hepatitis Associated with Teucrium Chamaedrys L. Decoction Use. Case Reports and Review of Literature. Basic Clin. Pharmacol. Toxicol. 109, 521–526 (2011).
Fox Ramos, A. E. et al. Revisiting Previously Investigated Plants: A Molecular Networking-Based Study of Geissospermum laeve. J. Nat. Prod. 80, 1007–1014 (2017).
Noor, H., Hammonds, P., Sutton, R. & Ashcroft, S. J. The hypoglycaemic and insulinotropic activity of Tinospora crispa: studies with human and rat islets and HIT-T15 B cells. Diabetologia. 32, 354–359 (1989).
Lokman, F. E. et al. Antidiabetic Effect of Oral Borapetol B Compound, Isolated from the Plant Tinospora crispa, by Stimulating Insulin Release. Evid. Based Complement. Alternat. Med. 2013, 1–7 (2013).
Ruan, C. T., Lam, S. H., Lee, S. S. & Su, M. J. Hypoglycemic action of borapetoside A from the plant Tinospora crispa in mice. Phytomedicine. 20, 667–675 (2013).
Noor, H. & Ashcroft, S. J. Pharmacological characterisation of the antihyperglycaemic properties of Tinospora crispa extract. J. Ethnopharmacol. 62, 7–13 (1998).
Noipha, K. & Ninla-Aesong, P. The Activation of GLUT1, AMPK alpha and PPAR gamma by Tinospora crispa in L6 Myotubes. Spatula DD. 1, 245–249 (2011).
Klangjareonchai, T. & Roongpisuthipong, C. The effect of Tinospora crispa on serum glucose and insulin levels in patients with type 2 diabetes mellitus. J. Biomed. Biotechnol. 2012, 1–4 (2011).
Pluskal, T., Castillo, S., Villar-Briones, A. & Orešič, M. MZmine 2: modular-framework for processing, visualizing, and analyzing mass spectrometry-based molecular data. BMC Bioinformatics. 11, 395–105 (2010).
Frank, A. M. et al. Clustering millions of tandem mass spectra. J. Proteome Res. 7, 113–122 (2008).
Acknowledgements
The authors would like to thank Dr Louis-Félix Nothias (CNRS) for his technical assistance and fruitful discussions and Valérie Dias for the English translation of our article.
Author information
Authors and Affiliations
Contributions
X.C., J.L. and D.B.M. wrote the main manuscript text. X.C., L.R.V., C.B., C.C., A.D. and S.M. developed the analytical techniques and participated in the analytical parts. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cachet, X., Langrand, J., Riffault-Valois, L. et al. Clerodane furanoditerpenoids as the probable cause of toxic hepatitis induced by Tinospora crispa. Sci Rep 8, 13520 (2018). https://doi.org/10.1038/s41598-018-31815-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-31815-6
This article is cited by
-
An updated and comprehensive review on the ethnomedicinal uses, phytochemistry, pharmacological activity and toxicological profile of Tinospora crispa (L.) Hook. f. & Thomson
Phytochemistry Reviews (2023)
-
Sexual-biased gene expression of olfactory-related genes in the antennae of Conogethes pinicolalis (Lepidoptera: Crambidae)
BMC Genomics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
