
Abstract
The arrangement of amino acids in a protein sequence encodes its native folding. However, the same arrangement in aggregation-prone regions may cause misfolding as a result of local environmental stress. Under normal physiological conditions, such regions congregate in the protein’s interior to avoid aggregation and attain the native fold. We have used solvent accessibility of aggregation patches (SAAPp) to determine the packing of aggregation-prone residues. Our results showed that SAAPp has low values for native crystal structures, consistent with protein folding as a mechanism to minimize the solvent accessibility of aggregation-prone residues. SAAPp also shows an average correlation of 0.76 with the global distance test (GDT) score on CASP12 template-based protein models. Using SAAPp scores and five structural features, a random forest machine learning quality assessment tool, SAAP-QA, showed 2.32 average GDT loss between best model predicted and actual best based on GDT score on independent CASP test data, with the ability to discriminate native-like folds having an AUC of 0.94. Overall, the Pearson correlation coefficient (PCC) between true and predicted GDT scores on independent CASP data was 0.86 while on the external CAMEO dataset, comprising high quality protein structures, PCC and average GDT loss were 0.71 and 4.46 respectively. SAAP-QA can be used to detect the quality of models and iteratively improve them to native or near-native structures.
Similar content being viewed by others


Introduction
The folding of a protein is a self-assembly process where the information of three dimensional (3D) structure is cryptically encoded in the primary sequence1. Successful prediction of a protein’s 3D structure from its primary sequence has been considered as a grand challenge in modern biology2,3,4. Protein folding involves a deep insight of the protein folding pathway, involving several intermediates5,6. Exhaustive sampling of all possible conformations is not a feasible theoretical solution for protein structure prediction (PSP) due to the large degrees of freedom available for proteins7. The dedicated pathway of protein folding is therefore via the thermodynamic folding hypothesis8, with the native state of a protein considered its most stable thermodynamic conformation. The hydrophobic effect is known to be a principal factor in the thermodynamic protein folding hypothesis, in addition to electrostatic interactions and conformational entropy9. Clustering of hydrophobic groups in a polar solvent is an “entropy-driven” process, which leads to the collapse of side chains to functional native conformations. This “hydrophobic collapse” is considered as the most popular protein folding model10,11,12. In contrast to protein folding which leads to the native state, protein misfolding is also a self-assembly process that results in an aggregated form. The same protein sequence can undergo folding or misfolding, depending on the physiological environment13. In the crowded cellular environment, there is always a possible chance for the protein to move in the direction of misfolding, suggesting that protein misfolding information is also encoded in its primary sequence.
The primary sequence of proteins may have several aggregation patches that are responsible for the formation of fibrils or amyloids. However, under physiological conditions, these patches self-assemble in the core of globular structures8,14 ruling out misfolding or aggregation and leading to the native structures. Aggregation-prone regions can be detected from protein sequences using several prediction programs15,16,17,18 and from protein structures19.
In this study, we have studied the packing and spatial positions of these ‘aggregation-prone’ patches in the native and non-native states of proteins. We developed a hypothesis on solvent accessibility of aggregation patch (SAAP), based on the first (http://predictioncenter.org/casp12/target.cgi?id=3view=all) CASP12 target20, which was applied to 1557 native structures from the Protein Databank (PDB)21 for validation. We then used the predicted structures from the CASP 12 homology model dataset to test our hypothesis, by comparing SAAP with the global distance total (GDT) score, which measures the deviation of the model from the experimental 3D structure. We observed that SAAP decreases exponentially as we move from non-native to native folds. Here, we have used folds in the context of structural domains, rather than complete protein structures which could comprise several domains. Our results support folding as a preferred pathway for globular proteins, accompanied by burying aggregation-prone residues from the solvent in their native states while these residues are more exposed in their non-native states or in aggregates. Thus, the SAAP score for the entire protein fold, SAAPp provides a direct structural metric to identify near native folds from misfolded structures. Moreover, minimizing SAAPp at an early stage of structure prediction can filter out non-native states, opening a new avenue for improved protein structure prediction. Quality assessment of predicted protein structure is broadly classified into single model quality assessment22,23,24,25,26,27,28,29,30 and consensus model quality assessment31,32,33. These scoring functions have been used to detect the quality of predicted protein models. Therefore, SAAPp was trained using a machine learning method (random forest) to evaluate the quality of these models applying the protein folding with aggregate formation paradox. The scoring function developed using SAAPp, SAAP-QA, showed excellent results comparable with the state-of-art methods in this field.
Results
We calculated the SAAPp score for the target ‘T0859’ from CASP12 as a case study, followed by validation on 1557 PDB native structures. Our hypothesis that SAAPp is a measure of protein folding was then tested on CASP12 TBM (template based model) predictions. Based on the results obtained, the SAAPp score was formulated into a scoring function, SAAP-QA, using random forest machine learning approach for evaluating the quality of protein models. Here 10978 CASP11 and CASP12 TBM models were used as training and test sets, with the remaining 4305 CASP12 models forming the blind test set. Furthermore, we validated SAAP-QA additionally on 51 targets from the CAMEO platform34.
Aggregation patches in T0859-a case study
In order to demonstrate the concept of minimum solvent accessibility of aggregation patches, the first CASP12 protein target T0859 was chosen as a case study. This is the Acinetobacter phage 205 (AP205) coat protein of 133 amino acids. Initially, the complete primary sequence of this protein was submitted to the Aggrescan server35 to predict aggregation-prone regions in the polypeptide sequence. The Aggrescan server uses aggregation propensity values per amino acid derived from experimental data, change in hydrophobicity, β-sheet propensity and the charge of the protein35. A window size that depends on the protein length is selected to calculate average aggregation propensity and the resultant value is assigned to the central residue as its aggregation value. Aggregation-prone “hot spot” patches have a high propensity to nucleate and initiate the aggregation process when exposed to a polar solvent. Figure 1a shows the primary sequence of the selected protein, T0859 with the corresponding aggregation-prone residues shown in red. These nine hot spot regions constitute 62 residues. Thus, 47% region of this protein has been predicted as aggregation-prone at sequence level. Many predicted aggregation-prone regions are shielded because they are buried in the protein’s hydrophobic core or involved in non-covalent interactions at the protein secondary and tertiary structural levels. Further, native structure of 131 residues (missing residues 1 and 2) of T0859 (PDB code: 5JZR) was submitted to the Aggrescan-3D (A3D) server19 to detect aggregation-prone residues for the given structure. The residue-wise scores are shown in Fig. 1c, with residues having positive scores considered aggregation-prone. The aggregation propensity of the same amino acids in the native 3D structure showed a reduction in aggregation-prone patches compared to the sequence-based prediction, due to protein folding. The A3D server detected 27 aggregation-prone residues out of 131 that constitute 21% of the complete structure. The results indicate that 35 residues having a high propensity to participate in aggregation from sequence-based analysis, this propensity was diminished in the native structure. In summary, structure-based prediction lowered aggregation-prone regions by 55.3% (47-21/47) in the native structure, from the sequence-based method. To illustrate further, aggregation prone patches on 3D structure of native and non-native conformations of AP205 are shown in Fig. 1d. It shows that non-native has bigger area for aggregation-prone regions than native. As the A3D server is only accessible via RESTful URIs, and is therefore unsuited for local installation, large-scale structure analysis using this method is unfeasible. Moreover, A3D score has major contribution of solvent exposed surface of individual residues. We therefore determined the spatial location of all AP205 residues, by calculating their solvent accessible surface areas (SASA; described in the Methods section), using a local copy of the naccess program36. The side chain accessible surface area for each residue is used as a marker to represent the exposure of any given residue to the solvent. Residues that showed greater than 50% of side chain solvent accessible surface area (SCsasai; described in the Methods section) are considered surface exposed residues, compared to earlier studies with a minimum cut-off 20% for solvent exposure37,38. Figure 1b shows the residue-wise solvent accessibility from naccess and aggregation propensity predicted by Aggrescan. Residues with SCsasai > 50% are given a score of ‘1’ and considered as surface accessible while all others are classified inaccessible and marked as ‘0’. As noted earlier, 62 residues (in red) are predicted as aggregation-prone using sequence-based prediction, and 27 of these (highlighted in blue) are solvent accessible as shown in Fig. 1c, consistent with A3D results. The reduction in solvent accessibility of aggregation-prone residues by 21% in their native structure suggests close packing of these residues in the interior of the protein. Figure 1a–c collectively demonstrates reduction of aggregation propensity as we move from primary sequence to structure. This can be quantified as solvent accessibility of aggregation patches (SAAPp) score (as defined in Eq. 1 in the Method section), computed as 43.5 for AP205 protein. Thus, SASA of predicted aggregation-prone patches may act as marker of their 3D location. The arrangement of aggregation-prone residues in tertiary structures could be perceived as a strong driving force for native protein folding.

Aggregation-prone regions. (a) Aggregation-prone regions predicted by Aggrescan server for the CASP 12 target T0859, highlighted in red. (b) Solvent accessibility of aggregation-prone residues predicted from side chain solvent accessible surface area (SASA) and aggregation propensity. (c) Individual aggregation score for each residue predicted by the A3D server, where positive scores correspond to aggregation. (d) Frequency of loss in SASA of aggregation-prone residues on native protein structures collected from PDB, as measured by SAAPp scores. (e) SAAPp scores mapped to the 3D surface of the native T0859 structure and its corresponding decoy, coloured by predicted aggregation propensity from red (highly aggregation-prone) to blue (least aggregation-prone or hydrophilic).
Validation on native crystal structures
As shown in the case study, the native protein structure leads to considerable loss in solvent accessibility for aggregation-prone residues. In order to examine the universality of this phenomenon, native crystal structures from the PDB database were analyzed. Aggregation-prone residues are predicted using the Aggrescan server for their primary sequences and their corresponding SASA was calculated using the naccess program. The SAAPp score was computed as per Eq. 1 (see Methods section). SAAPp score is intrinsically normalized with respect to the number of aggregation-prone residues in the polypeptide, to address the amino acid length of different native proteins. Among the 1557 selected native structures, 51 did not show any aggregation patches in their polypeptide sequence. Therefore, SAAPp was calculated for the remaining 1505 structures. Figure 1d shows the frequency distribution curve of these structures for different ranges of SAAPp. From the plot, 1358 structures have SAAPp ≤ 30, i.e. 90% of native structures have only 30% predicted aggregation-prone residues as solvent-exposed. This distribution confirms that native structures tend to move their aggregation-prone residues into the core, in order to reduce their solvent accessibility. In this calculation. 10 proteins with SAAPp > 50 are either aggregation-prone (coiled coil: 1M5I, 3K29, 3QFL, 5APZ and 5VO5), multimeric (trimeric: 2WB3, 2WH7, 3EMI and 3WP8) or phosphoinositide-binding (2WWE).
SAAP as a measure for CASP12 models
We then applied this concept of non-native/decoy structures being unfolded and thus aggregation-prone, to investigate the potential of SAAPp as a structural metric to detect the quality of predicted models for the CASP12 template based model (TBM) category. These predicted models contains near native structures in pools of ‘decoys’. Targets selected under the CASP12 TBM category with their different domains, are shown in Table 1, comprising 37 structural domains. Of these, 30 were selected, as five domains are small (<100 amino acids in length) and therefore have no significant hydrophobic core, while another two domains have very small aggregation-prone patches (<20%) of their complete sequence. The best models predicted by different servers were taken with their corresponding GDT (global distance total) scores, resulting in 6149 models from 30 target domains. Aggregation-prone residues for each target sequence were predicted followed by SAAPp calculation for each model structure. Inferences drawn from the case study and their extension to native crystal structures suggest that low SAAPp scores are representative of native or near native structures. Plotting SAAPp against GDT gives an exponential relation from curve fitting. (y = e(a + bX)) as shown in Supplementary Figure S1. Overall, the SAAPp score decreases from high quality models (near native) to poorly predicted models. Native and near native structures have the least SAAPp scores, maximizing their chance of attaining the native state. Each plot in Supplementary Figure S1 shows the top 10 models in red colour as per their GDT score. It can be seen that the top ten models can be detected using SAAPp scores. Pearson correlation coefficient (PCC) values between GDT and SAAPp scores are shown for each target in Table 2 with the sequence length of the model domain, to provide some indication of the globularity of the protein fold. The average PCC for 30 models (Table 2) is 0.76, where the maximum is 0.86 for T0910-D1 targets and the minimum is 0.52 for T0911-D1. These correlations clearly suggest that SAAPp can be used as structural metric to rank predicted protein structural models in the absence of their native structure. There is no observed correlation found between PCC values and the length of the proteins. A total of 3996 models out of 6149 have GDT > 50 where 99.7% of them showed SAAPp ≤ 50. This shows that the GDT cut-off score of 50 for SAAPp can be used to screen good models from set of decoys. Similarly, of 786 high quality models (GDT > 80), 89.4% have SAAPp < 40. This again confirms the possible application of SAAPp in screening high quality models from poor predictions.
SAAP Based Scoring Function, SAAP-QA
SAAPp has the potential to be developed further, as a scoring function for quality assessment of protein structure models by predicting GDT score of a given protein model. Machine learning approaches have been extensively used to build quality assessment scoring functions39,40,41,42. This requires strong descriptors/features for representing the quality of models. For 30 CASP12 target domains, SAAPp correlates with GDT with an average PCC of 0.76, suggesting its importance as a major descriptor in building a scoring function. In addition to SAAPp other physico-chemical descriptors were added to build a robust universal scoring function. The random forest43,44 machine learning method is used to for formulating a decision based prediction algorithm to predict the GDT score using input descriptors. Protein are structurally diverse in nature, therefore physico-chemical descriptors of proteins may not follow any strict rules to represent structural folds. In such a case, any linear or logistic regression approach would not fit the prediction method. Random forest is a decision tree method and it is highly applicable when individual descriptors have diversity as well as belong to multiple classes. Earlier methods in protein quality assessment used support vector machine (SVM)33,45 this technique is intrinsically suited for binary class problem. Protein quality assessment cannot be purely represented as binary problem (good vs bad models) as it is an example of multiclass problem where models span across continuous quality spectrum. Random forest (RF) is a fast machine learning method as one of the most powerful scalable and interpretable prediction model. In addition to this, RF is equally competent for classification and regression problem. Overfitting can result in biased prediction model specially when the dataset size is moderate and RF is less prone to overfitting/overtraining than support vector machine (SVM) or neural network (NN). SVMs are also best designed for a binary classification problem. These reasons made random forest more suitable for building the SAAPp scoring function, SAAP-QA.
Physico-chemical Descriptors for SAAP-QA
Here, SAAPp is used as major descriptor to build a scoring function for quality assessment. In addition, other descriptors that influence the SAAPp score are also included to build a robust scoring technique. TBM category models from CASP11 and CASP12 are used as dataset for training and testing. This dataset comprises 10978 models with 9135 from CASP11 and 1843 models from CASP12. Moreover, 4305 models from CASP12 are additionally used for blind testing. These models belong to different domains/targets, with varied secondary structure (helix, sheet and loop) composition. Residues involved in these secondary structural elements differ in their propensity for solvent accessibility. In order to account for this effect, helix, sheet and loop fractions were used as descriptors in addition to the SAAPp score. Moreover, loop residues show high degrees of freedom in the tertiary structure of proteins and are therefore more susceptible to solvent accessibility changes. This attribute makes the loop element another critical component that influences the SAAPp score. To provide a higher weightage to loops in scoring function, the total loop content and loop to SAAPp ratio were added in the descriptor list. In summary, six descriptors (detailed in Method section) are used to build the scoring function: (1) SAAPp (2) helix fraction (3) sheet fraction (4) loop fraction (5) loop content and (6) SAAPp/loop ratio. Density plot and individual correlation for all six descriptors for CASP11 and CASP12 models are shown in Supplementary Figure S2. As expected, SAAPp has highest correlation with GDT (PCC = 0.57) followed by loop fraction (PCC = 0.47) and then the helix fraction (PCC = 0.27). Supplementary Figure S2 shows, SAAPp, loop fraction and total loop content follow normal distribution pattern for the dataset with bell shaped curve while the SAAPp/loop ratio has skewed distribution. Helix and sheet fraction are multimodal distribution with more than one peak value. The GDT distribution also shown in Supplementary Figure S2, shows a multimodal pattern with two major peaks at values ‘13’ and ‘75’. From Supplementary Figure S2, the six descriptors that were selected to build a scoring function using RF machine learning method, showed uniform distributions on the combined CASP11 and CASP12 dataset. These six are distinct in nature, contributing individually to GDT score prediction with maximum PCC of 0.71 with SAAPp (shown in Supplementary Figure S3). Further, after the data is divided into training, test and blind test sets, the individual correlation of these features with their GDT was re-calculated. Here again, SAAPp has maximum PCC of 0.57, 0.58 and 0.64 with GDT while SAAPp/loop has low individual PCC of 0.28, 0.26 and 0.15 with GDT on training, test and blind datasets respectively.
Training and Test Dataset Performance (CASP11 and CASP12 Targets)
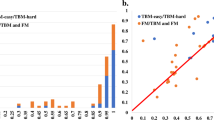
Structures from CASP11 and CASP12 are used as datasets for building the prediction model, with 53 unique targets divided randomly into training and test sets, comprising fractions of 70% and 30% respectively. T0852 has two domains so it was counted once. The resulted in 38 targets consisting of 7907 protein models in the train set and 15 targets with 3071 protein models in the test set. These targets are listed in Supplementary Table S1 and their division into train and test sets is shown in Supplementary Table S2. As the models are randomly split into train and test sets based on targets, the two datasets do not have any common target protein, making the learning process unbiased. A complete list of models with their individual GDT and the six features that were used in prediction model building are shown in Supplementary Table S2. The prediction model showed a high correlation between observed and predicted GDT on train and test set with Pearson correlation coefficient (PCC) values of 0.96 and 0.86 respectively. Figure 2 shows the relation between predicted GDT using SAAP-QA and the other descriptors with the true GDT score for the training and the test sets. Structural metrics for accessing the quality of models are evaluated on their classification ability characterizing good and bad models. SAAP-QA was tested on this parameter using receiver operating characteristic (ROC) values, with GDT cut-off set as 50 based on earlier study46. Hence, models with GDT > 50 score are considered as high quality models while GDT < 50 were considered as low quality models. True positive and false positive rates (TPR and FPR) on different cut-off values of predicted GDT were calculated and plotted to determine its ability to categorize good and bad models. Figure 3(a) shows ROC curves for training and test datasets have good classification for all targets. The area under the curve (AUC) for each ROC was computed as 0.98 and 0.94 for training and test sets, respectively (shown as label in Fig. 3). Figures 2 and 3(a) collectively show the high performance of SAAP-QA as a structural scoring metric to rank protein models and for further classifying them as good and poor quality models. Furthermore, 3-fold cross validation was performed to assess how the results of prediction model could be generalized to an independent set. Here, the complete data set was randomly divided into three groups based on targets having 18 (3812 protein models), 18 (3869 protein models) and 17 (3297 protein models) targets respectively. The complete list of these three sets used in cross validation is given in Supplementary Table S2, again with none of the sets having any common target and thus comprised of distinct protein models. In each run, two sets were used to train the machine learning model while the third independent set was used for testing. The average AUC of 3-fold cross validation is 0.93 while PCC is 0.83 as shown in Fig. 3(b). Consistent AUC and PCC values with standard deviation values (σ) of 0.007 and 0.002, respectively on the three distinct cross-validation datasets confirms the robustness of prediction model.

Evaluation of GDT predicted by SAAP-QA on CASP11/CASP12 train and test set. Compared to true GDT values, SAAP-QA predicted GDT values show correlation coefficient values (r) of (a) 0.96 for the training set with with 38 targets consisting of 7907 models and (b) 0.86 for the test set with 15 targets with 3071 models.

SAAP-QA performance on train and test set from CAP11/CASP12. (a) Classification of good and bad models representing the ROC curve for training set (38 targets with 7907 models) with an AUC of 0.98 and for test set (15 targets with 3071 models) with an AUC of 0.94. (b) Shows correlation coefficients (PCCs) and area under the curve (AUC) for 3-fold validation test of prediction model, average PCC for three distinct dataset is 0.83 while average AUC is 0.93. Standard deviation of these 3 runs are 0.007 and 0.002 for PCC and AUC respectively.
Training and Test Set Per Target Evaluation
Top model for each target is selected based on the prediction made by SAAP-QA. The GDT loss for each target is calculated as the difference between the GDT score of best model selected by SAAP-QA with the best GDT score model available in the decoy set, as shown in Supplementary Table S3. This table shows that the average GDT loss on train set for 38 targets is 0.80 while it is 2.02 on the test set for 15 targets. Pearson correlation co-efficient (PCC) between predicted GDT and actual GDT for each target is calculated as shown in Supplementary Table S4. The average PCC for the train set is 0.96 while for the test set, the average PCC is 0.86.
Blind Test Set Performance (CASP12 and CAMEO)
CAMEO dataset is a repository of high quality models. Here, models are deposited more frequently than CASP experiment but the number of models for each target is lesser than CASP. A dataset of 51 targets from the CAMEO platform used in the blind test set comprise 1489 models, as shown in Supplementary Table S5. These structures are completely unknown for the prediction model as they are not from train/test set. Similarly, 18 CASP12 targets were also excluded from training and test sets for further use of blind testing. Although the number of targets for the blind set from CASP12 is 18 but it comprises of 4305 models, which is larger than the prediction model test set of 3071 models. CASP12 models considered in the blind test is shown in the Supplementary Table S5 with their corresponding six physico-chemical features used in building the SAAP-QA and their respective GDT score. In summary, 5794 models were used in the blind test set from 69 targets (CAMEO + CASP). Further, CASP12 targets were separately tested for domains and full protein structure.
CAMEO-Complete Structures
Continuous Automated Model Evaluation (CAMEO) is a continuous blind prediction assessment for protein structures which are going to be published in the subsequent weekly release of PDB34. This platform releases targets every week. For validation of SAAP-QA, recently submitted models were collected from CAMEO. This set consisted of 61 targets. Four targets less than 50 residues in length while six are above 500. These 10 targets were not considered in the study, being too small to have a hydrophobic core or multi-domain in nature. The selected 51 targets with their residue length are shown in Supplementary Table S6. The values of SAAPp, helix fraction, sheet fraction, loop fraction, loop content and SAAPp/loop ratio were calculated for each model for all 51 targets. Further, SAAP-QA was used to predict the GDT using these six physico-chemical features. Top 1 model was selected for each target using the predicted GDT score. Individual GDT loss of the selected model with the best available model was calculated to evaluate the performance of SAAP-QA on CAMEO targets. Further, GDT loss for top 5 and top 10 were also computed. Table 3 shows the list of 51 targets with respective GDT loss on top 10, top 5 and top 1 model selected by SAAP-QA. The average GDT loss for top 1 model for 51 targets is 4.46 while for top 5 and top 10, the average GDT loss is 2.12 and 1.01, respectively. Here, target 5VH2 and 5VO3 shown high GDT loss due to the extended sheet component in their 3D structure. Similarly, correlation coefficient was also calculated for 51 CAMEO targets between predicted and true GDT. Overall correlation coefficient (PCC) on CAMEO dataset is 0.71, with individual PCC values for each target is shown in Supplementary Table S4. Predicted GDT compared to true GDT is shown in Fig. 4(a). AUC value for CAMEO data was 0.82 as shown in Fig. 4(b).

SAAP-QA performance on CAMEO Set. (a) Comparison between predicted GDT by SAAP-QA and true GDT on CAMEO dataset showed overall correlation coefficient (PCC) 0.71 (b) classification of good and bad models representing the ROC curve for blind test on CAMEO models (51 targets) with an AUC of 0.82.
CASP12 - Domain Structures
Single or multiple domains are predefined for each targets during CASP. These domains assist in detailed categorization of targets into (1) High Accuracy Modeling category - that will include domains where the majority of submitted models are of sufficient accuracy for detailed analysis and (2) Topology category (formerly Free Modeling) - that will assess domains where all submitted models are of relatively low accuracy. As SAAP-QA performs better on high quality models where high GDT score structures are available, we have considered specific domains of CASP targets instead of complete structures under the high accuracy modeling category for model development. Moreover, CASP itself gives weightage to these domains and reports domain-wise detailed quality analysis http://predictioncenter.org/casp12/results.cgi20. Implementation of a scoring function on the blind dataset of domain-wise models produces an overall correlation coefficient (PCC) of 0.76 between observed and predicted GDT (individual PCC for each target is shown in Supplementary Table S4). Based on CASP quality assessment (QA) results, the models generated by servers are classified into stage 1, which are closer to the experimental structure, based on their GDT values, and stage 2, which are the rest of the predicted structures. Stage 1 has 20 models for each target while stage 2 has 150 models selected by the Davis-QAconsensus method47. Stage 1 models for each target were ranked using SAAP-QA. The GDT loss of top ranked models for each target by SAAP-QA and the best available model in the pool are ‘0’ for stage 1 models. These results show that the SAAP-QA is able to capture the best model every time at first position in the decoy set for stage 1 models. As a next step, stage 2 models were also tested using SAAP-QA for ranking. Stage 2 is considered more important than stage 1 for structure prediction, and is used for QA method evaluation, as it is essential to eliminate model structures that are far from the experimental structure efficiently. For stage 2, 150 models were preselected by CASP organizers for quality assessment servers (QA). These 150 models for each of 18 target domains (listed in Table 4 as the blind test test) as ranked by different QA servers in the CASP12 competition, were evaluated using SAAP-QA. CASP allows the submission of the top 5 models in the 3D structure prediction category. Table 4 tabulates the GDT loss of the best model available in the pool with the top 5 and the top 10 captured by our scoring function. Average GDT loss for these targets in the top 5 models is 3.14 for stage 2 models, i.e. SAAP-QA selects the top 5 models that have 3.14 average GDT deviation from the best model available in the pool. Similarly, the top 10 models selected by SAAP-QA and their GDT loss were also shown in Table 4. The average GDT loss for the top 10 models is 1.72. Lastly, the top model (referred to as top 1) selected by SAAP-QA was also evaluated, with an average GDT loss of 6.41 (shown in the last column of Table 3). Data presented in Table 4 shows the ranking ability of SAAP-QA on an independent dataset. Individual PCC for each target in the blind test set between the predicted and actual GDT is shown in Supplementary Table S4 while GDT loss for the top 1 model is shown in Supplementary Table S3.
CASP12-Complete Structures
SAAP-QA is designed for structural domains but it can also be implemented for complete protein structures. In order to compare with CASP QA servers, we tested our prediction model on complete structures from “stage 2” models for 17 targets (T0920 is duplicated in domain study as it has two domains). Here, we calculated the GDT loss for top 10, top 5 and top 1 structures selected by SAAP-QA. For comparison with other scoring functions, the top three QA servers from CASP12 were selected. These servers, outperforming others during competition, are: (a) SMQA48 (b) ProQ349 and (c) MESHI_CON_SERVER50. The average GDT loss for these three servers on the selected 17 targets (with full structure) were calculated afresh. SVMQA has 3.8 average GDT loss while ProQ3 and MESHI_CON_SERVER have 3.69 and 5.14 GDT loss, respectively for the best predicted model from stage 2. Comparatively, SAAP-QA showed 6.08 average GDT loss for full protein structures on these 17 CASP12 targets, with the individual GDT loss for each target shown in Table 5. Moreover, SAAP-QA captured models among top 5 and top 10 categories with average GDT loss of 3.12 and 2.28, respectively. Detailed scores are shown in Table 5. SAAP-QA showed comprarable performance with the state-of-art QA servers, as well as efficiently capture native/near-native models in the top5/top10 bin. Thus, it can be integrated with protein structure prediction programs to screen high quality models. It should also be noted that although SAAP-QA was modeled on domains, it can be applied to full length protein structures.
Conclusions
The primary sequence of a protein has been considered to encode its native (or 3D) structure but it also encodes residue-level information about aggregation. Under standard physiological conditions, protein selects its native folding pathway and avoids aggregation. To the best of our knowledge, protein aggregation has focussed on understanding disease propensity including amyloid and fibril formation and has not been applied extensively to protein structure prediction to address the protein folding problem. Folding and the spatial conformation of aggregation-prone patches can help in solving the protein folding problem. Here, we have examined solvent accessibility of aggregation patches (SAAPp) on native and decoy structures. Native structures showed smaller SAAPp values, suggesting the close packing of these aggregation-prone residues in the core of their respective structures. However, non-native structures showed higher SAAPp values, indicating the exposure of a larger proportion of aggregation-prone residues to the solvent compared to native structures. CASP12 models under the TBM category were examined to uncover the relevance of SAAPp scores for predicted protein structures. The results showed a high overall correlation of 0.76 between SAAPp and GDT scores on 30 target domain structures of CASP12. Furthermore, SAAPp along with 5 other structural descriptors were trained using random forest machine learning approach to build the SAAPp scoring function, SAAP-QA. So as to add diversity in the data set, 21 CASP11 targets domain structures were also added to the 13 CASP12 targets during scoring function formulation. Train and test set was divided based on protein targets to avoid any similarity between train and test set protein models. SAAP-QA showed correlation co-efficient of 0.96 and 0.86 on train and test sets respectively while the average area under the ROC curve (AUC) values for distinguishing true positives from false positives was 0.98 and 0.94, respectively. Generalized effect of prediction model was tested using 3-fold validation. Average PCC in 3-fold validation was 0.83 while AUC was 0.93. Small standard deviation in PCC and AUC during 3-fold validation showed robustness of prediction model on independent dataset. Further, CAMEO was used as an external validation dataset for blind testing the prediction model, 51 targets from CAMEO platform were tested for this purpose. The average GDT loss for top 10, top 5 and top 1 models were 1.01, 2.12 and 4.46, respectively, for CAMEO targets. In addition to the 51 CAMEO targets, 18 CASP12 targets (both domains and full structures) were also added to the blind test set that were not part of model training/testing. The result showed 0.76 PCC between predicted and actual GDT. Ranking ability was further tested using GDT loss between models captured by SAAP-QA and the best model available. Average GDT loss on stage 2 (150 models) domains structures was computed as 6.08, 3.12 and 2.28 for top 1, top 5 and top 10 ranked models respectively. This combined showed its performance on an external unseen sample set. SAAPp is thus a computational measure of the degree of protein folding, which naturally tends to minimize the solvent accessible area for aggregation-prone residues. SAAPp has shown noteworthy performance in classifying good and bad models and can serve as an independent metric for separating near-native prediction model structures from poorly predicted model structures and for incorporation in protein structure prediction algorithms, to eliminate decoy structures and iteratively improve near-native models.
Methods
The first CASP12 target “T0859” from Acinetobacter phage AP205 (listed in the category Human and Server) was selected as a case study. High quality PDB structures were then selected, applying the criteria of: (1) number of chains = 1, (2) 100 ≤ sequence length ≤500; (3) resolution ≤ 2 Å; (4) type of macromolecule = only protein; (5) no ligands were present and (6) sequence identity ≤30%, resulting in 1557 structures. Test structures were used from CASP 12 automatic evaluation results for the template-based model (TBM) category predicted by different participating servers, comprising 37 domains from different targets where 5 domains are small (<100 amino acids in length) and therefore have no hydrophobic core while another 2 proteins have very small aggregation-prone patches (<20%) of their complete sequence. Thus 30 shortlisted targets are: T0860-D1, T0861-D1, T0867-D1, T0871-D1, T0873-D1, T0877-D1, T0879-D1, T0881-D1, T0883-D1, T0885-D1, T0889-D1, T0891-D1, T0893-D2, T0895-D1, T0902-D1, T0906-D1, T0910-D1, T0911-D1, T0912-D1, T0913-D1, T0917-D1, T0920-D1, T0920-D2, T0921-D1, T0928-D1, T0943-D2, T0944-D1, T0946-D2, T0947-D1 and T0948-D1. Here ‘D1/D2’ represents the domain name assigned by CASP organizer. Solvent accessible surface area (SASA) was calculated using the naccess program based on Lee and Richards’ algorithm51. The solvent accessibility of aggregation patches (SAAP) score for each protein p (SAAPp), was calculated from residue scores i, using side chain solvent accessible surface area (SCsasai) values and predicted aggregation patches (PREDAggregationi), as shown in Equation 1. SAAPp scores were then computed as the percentage solvent accessible aggregation-prone residues, as shown in Equation. 1.
SAAPp = SAAP score for protein ‘p’; SAAPi = SAAP score for residue ‘i’ AggreProneT = Total number of aggregation-prone residue
SCsasai is toal surface area for side chains of amino acids exposed to water solvent. Aggregation-prone regions were predicted consistently using the Aggrescan server35 for protein sequences and the Aggrescan3D (A3D)19 server for 3D structures. Multiple protein sequences were submitted to the the aggregation server available at http://bioinf.uab.es/aggrescan/http://bioinf.uab.es/aggrescan/ to calculate individual aagregation score of amino acids, Supplementary Figure S4 shows that Aggrescan server can handle multiple sequences and produce the result in very short time. However, Aggregation3D (A3D) server available at http://biocomp.chem.uw.edu.pl/A3D/http://biocomp.chem.uw.edu.pl/A3D/ used only once for a case study (CASP Target T0859), here 3D structure of protein was submitted to server that gives A3D scores for individual residues.
Random forest, a decision based machine learning approach was used to build SAAPp scoring function. This scoring function was designed to predict GDT of a given protein model using SAAPp and other descriptors. Multiple descriptors were used to build scoring function, SAAPp served as major descriptors. Additional descriptors along with SAAPp were added to build robust scoring function in order to capture diversity among protein’s structures. Following descriptors were used in machine learning method to build SAAPp scoring function: (A) SAAPp Score - calculation described above. (B) Helix fraction-ratio of total number of residues involved in helix formation to the length of protein. (C) Sheet fraction - ratio of total number of residues involved in sheet formation to the length of protein. (D) Loop fraction - ratio of total number of residues involved in loop formation to the length of protein. (E) Loop content-total number of residues involved in loop formation. (F) SAAPp/Loop -ratio of SAAP score with total number of residues involved in loop formation. Secondary structure of protein was assigned using STRIDE program, it implements a knowledge-based algorithm that makes combined use of hydrogen bond energy and statistically derived backbone torsional angle information52.
Model training was done using the ‘R’ package53 where 700 trees were grown in the forest using 2 features at every split. All protein Models from CASP11 and CASP12 were mixed, resulting in a total of 10978 models and 53 unique targets. These models were then randomly split into train (38 targets, 7907 models) and test (15 targets, 3071 models) sets based on targets following 70/30 rule. Although, data division is based on non-overlapping targets i.e. train set has 70% while test set has 30% unique targets, but the number of protein models in train and test set are also in 70% and 30% proportion. Later, these 53 targets divided into 3 non overlapping sets for conducting 3-fold validation. These sets have 18, 18 and 17 targets comprising 3812, 3869 and 3297 protein models respectively. In each run of 3-fold validation, any two sets were used for training while the third one was used as test set. All these data points belong to the CASP11 and CASP12 TBM category models and the complete list of the names of the targets used for training and testing is provided in Supplementary Table S1. In addition to training and testing, a blind validation was also performed to evaluate the performance of prediction algorithm on an unknown set. The blind validation dataset is comprised of 4305 models belong to 18 targets from CASP12 that were not used in building the prediction algorithm. These 18 cases were preselected from the total list of 30 and they are the last 18 TBM category targets. Furthermore, SAAPp scoring function, SAAP-QA, was also validated on CAMEO targets for blind prediction testing that has high quality models thn CASP experiment. Here, April 2018 targets were selected from the CAMEO website https://www.cameo3d.org/ https://www.cameo3d.org/. This constituted 51 targets after ignoring those with residue length less than 50 and more than 500. The GDT score was given for each model in its downloaded score file while the fasta sequence is provided separately with every target.
References
Anfinsen, C. B. Principles that govern the folding of protein chains. Sci. 181, 223–230 (1973).
Mushegian, A. Grand challenges in bioinformatics and computational biology. Front. genetics 2, 60 (2011).
Unger, R. & Moult, J. Finding the lowest free energy conformation of a protein is an np-hard problem: proof and implications. Bull. Math. Biol. 55, 1183–1198 (1993).
Berendsen, H. J. A glimpse of the holy grail? Sci. 282, 642–643 (1998).
Baldwin, R. L. The nature of protein folding pathways: the classical versus the new view. J. biomolecular NMR 5, 103–109 (1995).
Englander, S. W. & Mayne, L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. 111, 15873–15880 (2014).
Zwanzig, R., Szabo, A. & Bagchi, B. Levinthal’s paradox. Proc. Natl. Acad. Sci. 89, 20–22 (1992).
Bryngelson, J. D., Onuchic, J. N., Socci, N. D. & Wolynes, P. G. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins: Struct. Funct. Bioinforma. 21, 167–195 (1995).
Dyson, H. J., Wright, P. E. & Scheraga, H. A. The role of hydrophobic interactions in initiation and propagation of protein folding. Proc. Natl. Acad. Sci. 103, 13057–13061 (2006).
Lazar, G. A. & Handel, T. M. Hydrophobic core packing and protein design. Curr. opinion chemical biology 2, 675–679 (1998).
Ponnuswamy, P., Prabhakaran, M. & Manavalan, P. Hydrophobic packing and spatial arrangement of amino acid residues in globular proteins. Biochimica et Biophys. Acta (BBA)-Protein Struct. 623, 301–316 (1980).
Zhu, B.-Y., Zhou, M. E., Kay, C. M. & Hodges, R. S. Packing and hydrophobicity effects on protein folding and stability: effects of b-branched amino acids, valine and isoleucine, on the formation and stability of two-stranded a-helical coiled coils/leucine zippers. Protein Sci. 2, 383–394 (1993).
Chiti, F. et al. Kinetic partitioning of protein folding and aggregation. Nat. Struct. Mol. Biol. 9, 137 (2002).
Dobson, C. M. Protein folding and disease: a view from the first horizon symposium. Nat. Rev. Drug Discov. 2, 154 (2003).
Fernandez-Escamilla, A.-M., Rousseau, F., Schymkowitz, J. & Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. biotechnology 22, 1302 (2004).
Pawar, A. P. et al. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. molecular biology 350, 379–392 (2005).
Tartaglia, G. G., Cavalli, A., Pellarin, R. & Caflisch, A. Prediction of aggregation rate and aggregation-prone segments in polypeptide sequences. Protein Sci. 14, 2723–2734 (2005).
Trovato, A., Seno, F. & Tosatto, S. C. The pasta server for protein aggregation prediction. Protein Eng. Des. & Sel. 20, 521–523 (2007).
Zambrano, R. et al. Aggrescan3d (a3d): server for prediction of aggregation properties of protein structures. Nucleic acids research 43, W306–W313 (2015).
Moult, J., Fidelis, K., Kryshtafovych, A., Schwede, T. & Tramontano, A. Critical assessment of methods of protein structure prediction (casp)—round xii. Proteins: Structure, Function, and Bioinformatics 86, 7–15 (2018).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res 28, 235–42 (2000).
Mishra, A., Rao, S., Mittal, A. & Jayaram, B. Capturing native/native like structures with a physico-chemical metric (pcsm) in protein folding. Biochimica et Biophys. Acta (BBA)-Proteins Proteomics 1834, 1520–1531 (2013).
Mishra, A., Rana, P. S., Mittal, A. & Jayaram, B. D2n: Distance to the native. Biochimica et Biophys. Acta (BBA)-Proteins Proteomics 1844, 1798–1807 (2014).
Cao, R. & Cheng, J. Protein single-model quality assessment by feature-based probability density functions. Sci. reports 6, 23990 (2016).
Cao, R., Wang, Z. & Cheng, J. Designing and evaluating the multicom protein local and global model quality prediction methods in the casp10 experiment. BMC structural biology 14, 13 (2014).
Cao, R., Wang, Z., Wang, Y. & Cheng, J. Smoq: a tool for predicting the absolute residue-specific quality of a single protein model with support vector machines. BMC Bioinformatics 15, 120 (2014).
Park, J. & Saitou, K. Rotas: a rotamer-dependent, atomic statistical potential for assessment and prediction of protein structures. BMC Bioinformatics 15, 307 (2014).
Rykunov, D. & Fiser, A. Effects of amino acid composition, finite size of proteins, and sparse statistics on distancedependent statistical pair potentials. Proteins: Structure, Function, and Bioinformatics 67, 559–568 (2007).
Shen, M.-y & Sali, A. Statistical potential for assessment and prediction of protein structures. Protein science 15, 2507–2524 (2006).
Zhang, J. & Zhang, Y. A novel side-chain orientation dependent potential derived from random-walk reference state for protein fold selection and structure prediction. PloS one 5, e15386 (2010).
McGuffin, L. J. The modfold server for the quality assessment of protein structural models. Bioinforma. 24, 586–587 (2008).
McGuffin, L. J. & Roche, D. B. Rapid model quality assessment for protein structure predictions using the comparison of multiple models without structural alignments. Bioinforma. 26, 182–188 (2009).
Wang, Q., Vantasin, K., Xu, D. & Shang, Y. Mufold-wqa: A new selective consensus method for quality assessment in protein structure prediction. Proteins: Structure, Function, and Bioinformatics 79, 185–195 (2011).
Haas, J. et al. Continuous automated model evaluation (cameo) complementing the critical assessment of structure prediction in casp12. Proteins: Structure, Function, and Bioinformatics 86, 387–398 (2018).
Conchillo-Solé, O. et al. Aggrescan: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics 8, 65 (2007).
Hubbard, S. J. & Thornton, J. M. ‘NACCESS’, computer program. Department of Biochemistry and Molecular Biology, University College, London (1993).
Chen, H. & Zhou, H.-X. Prediction of solvent accessibility and sites of deleterious mutations from protein sequence. Nucleic acids research 33, 3193–3199 (2005).
Zhang, H. et al. On the relation between residue flexibility and local solvent accessibility in proteins. Proteins: Structure, Function, and Bioinformatics 76, 617–636 (2009).
Benkert, P., Künzli, M. & Schwede, T. Qmean server for protein model quality estimation. Nucleic acids research 37, W510–W514 (2009).
Hua, S. & Sun, Z. A novel method of protein secondary structure prediction with high segment overlap measure: support vector machine approach1. J. molecular biology 308, 397–407 (2001).
Ray, A., Lindahl, E. & Wallner, B. Improved model quality assessment using proq2. BMC Bioinformatics 13, 224 (2012).
Wang, Z., Eickholt, J. & Cheng, J. Apollo: a quality assessment service for single and multiple protein models. Bioinforma. 27, 1715–1716 (2011).
Breiman, L. Random forests. Mach. learning 45, 5–32 (2001).
Breiman, L. Prediction games and arcing algorithms. Neural computation 11, 1493–1517 (1999).
Jo, T. & Cheng, J. Improving protein fold recognition by random forest. BMC Bioinformatics 15, S14 (2014).
Maghrabi, A. H. & McGuffin, L. J. Modfold6: an accurate web server for the global and local quality estimation of 3d protein models. Nucleic acids research 45, W416–W421 (2017).
Schwede, T. et al. Outcome of a workshop on applications of protein models in biomedical research. Struct. 17, 151–159 (2009).
Manavalan, B. & Lee, J. Svmqa: support–vector-machine-based protein single-model quality assessment. Bioinforma. 33, 2496–2503 (2017).
Uziela, K., Shu, N., Wallner, B. & Elofsson, A. Proq3: Improved model quality assessments using rosetta energy terms. Sci. reports 6, 33509 (2016).
Kryshtafovych, A. et al. Assessment of the assessment: evaluation of the model quality estimates in casp10. Proteins: Structure, Function, and Bioinformatics 82, 112–126 (2014).
Lee, B. & Richards, F. M. The interpretation of protein structures: estimation of static accessibility. J. molecular biology 55, 3479–IN4 (1971).
Heinig, M. & Frishman, D. Stride: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic acids research 32, W500–W502 (2004).
R Core Team, R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, http://www.R-project.org/ (2013).
Acknowledgements
A.M. acknowledges the Department of Biotechnology (DBT), India for the award of an Indo-Australian Gold fellowship and support from the Institute for Integrated and Intelligent Systems, Griffith University. Authors acknowledge the support provided by Australian Reseach Council (Grant: DP180102727). The authors also thank the anonymous reviewers for their critical comments.
Author information
Authors and Affiliations
Contributions
A.M. conceived and carried out the analysis. A.M., S.R. and A.S. designed the work. A.M. and S.R. wrote the manuscript. All authors reviewed and agreed to the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mishra, A., Ranganathan, S., Jayaram, B. et al. Role of solvent accessibility for aggregation-prone patches in protein folding. Sci Rep 8, 12896 (2018). https://doi.org/10.1038/s41598-018-31289-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-31289-6
This article is cited by
-
WGS-based screening of the co-chaperone protein DjlA-induced curved DNA binding protein A (CbpA) from a new multidrug-resistant zoonotic mastitis-causing Klebsiella pneumoniae strain: a novel molecular target of selective flavonoids
Molecular Diversity (2023)
-
Exploring the sequence features determining amyloidosis in human antibody light chains
Scientific Reports (2021)
-
Protein aggregation: in silico algorithms and applications
Biophysical Reviews (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
