
Abstract
Vitiligo is a skin disease that affects 1% of the population worldwide. Both environmental and genetic factors contribute to the risk of vitiligo. GZMB encodes the enzyme Granzyme B, which plays an important role in cytotoxic T cell-induced apoptosis, and it has been considered one of the candidate genes for vitiligo because of its connections with human immune system. Overall, 3,120 study subjects with Chinese Han ancestry were recruited, and 15 pre-selected SNPs of GZMB were genotyped. Genetic association analyses were performed to evaluate the genetic risk of these SNPs to vitiligo. Further bioinformatic analyses were conducted to examine the potential biological function of targeted SNPs. The SNP rs8192917, a non-synonymous coding SNP, was identified to be significantly associated with the disease status of vitiligo, with OR = 1.39 and P = 1.92 × 10−8. Differences in the association signal can be observed in the stratification analyses of multiple clinical variables. Our positive results provide additional supportive evidence that GZMB gene is an important locus for vitiligo in Han Chinese population.
Similar content being viewed by others

Introduction
Vitiligo is a skin disease characterized by the loss of pigment in patches of skin1. Approximately 1% of the world’s population is affected by vitiligo, and in some countries, this percent can be as high as 2–3%2. In general, there is no significant difference in gender for susceptibility to vitiligo. Approximately half of vitiligo patients develop this disorder before 20 years old, and the age of onset of vitiligo for most patients is before 401. Currently, there is no cure for vitiligo but several treatment options can relieve the symptoms1. In addition, vitiligo patients may experience depression and relevant mood disorders due to the potential for discrimination from society.
Multiple hypotheses have been proposed for the etiology of vitiligo, and changes in the human immune system are considered to be among the most important causes1. Previous studies have shown that vitiligo is a complex disorder that is influenced by both environmental and genetic factors. Multiple genes contributed to the onset of this disease3,4; the heritability was approximately 46–72%5,6. In recent decades, candidate gene-based association studies have successfully mapped susceptibility for many complex diseases7,8,9,10,11,12,13. Genome-wide association studies have found multiple loci that contribute to the susceptibility of vitiligo. 48 loci have been reported in Caucasians, and a number more in Han Chinese14,15,16. Despite these findings, these loci explain only approximately 25% of the genetic risk of developing vitiligo. More research is needed to fully unravel the genetic mechanisms of vitiligo17,18,19.
GZMB is a protein coding gene that is located at 14q12 and has 5 exons, with a length of 3320 bp. The protein product of GZMB is an enzyme (Granzyme B) that plays an important role in the process of apoptosis induced by cytotoxic T cells20,21. In a recent GWAS study conducted by Jin et al. examining European populations, the SNP rs8192917 in GZMB was found to be significantly associated with vitiligo16. However, this finding has not been replicated in other populations. In this study, we aimed to investigate the potential association between polymorphisms of GZMB and vitiligo using 3,120 study subjects with Chinese Han ancestry. Including rs8192917, a total of 15 SNPs were selected for genotyping. The biological functions of targeted SNPs were examined further by bioinformatic analyses. Our results would provide clues for understanding the roles of GZMB in the genetic predisposition of vitiligo.
Methods
Study Subjects
In this study, 973 unrelated patients with vitiligo and 2,147 age- and gender-matched unrelated controls were recruited from the dermatological department of the Second Affiliated Hospital of Xi’an Jiaotong University. We only included Han Chinese patients who were born in the local area in an effort to have a genetically homogenous cohort of individuals. None of the patients had been subjected to any therapy in the 6 months prior to sampling. None of the healthy subjects showed any clinical evidence or family history of vitiligo or of any other autoimmune disorder. Vitiligo was clinically characterized in patients as segmental and non-segmental. Segmental vitiligo was diagnosed if the disease followed a dermatomal distribution, which involves one segment of the skin and shows early hair whitening and rapid progression. Active vitiligo was defined as the appearance of new lesions or the enlargement of existing lesions in the 3 months before presentation. Written informed consent was obtained from each subject. This research was performed in accordance with the ethical guidelines of the Declaration of Helsinki (version 2002) and was approved by the Ethics Committee of Xi’an Jiaotong University. The characteristic information of the study subjects is summarized in Table 1. No significant differences in distribution in cases and controls were identified for the age or gender of the study subjects.
SNP Selection and Genotyping
SNPs with a minor allele frequency (MAF) >0.01, heterozygosity >0.2 and located within the GZMB gene region were extracted for genotyping based on the 1000 genome CHB data. Overall, 15 SNPs were obtained. Genomic DNA was extracted from peripheral blood leukocytes according to the manufacturer’s protocol (Genomic DNA kit, Axygen Scientific Inc., CA, USA). Genotyping was performed for all SNPs using the MassARRAY platform (Sequenom, San Diego, CA, USA). The genotyping results were generated and processed by using Typer Analyzer software (Sequenom)22. The final genotyping call rate for each SNP was greater than 99%, and the overall genotyping call rate was 99.9%. The quality of our genotyping results ensured the reliability of further statistical analyses.
Statistical analyses
MAFs were calculated and Hardy-Weinberg equilibriums were tested for each SNP. Logistic regressions were performed for each SNP to evaluate their potential contributions to the risk of vitiligo. The potential inflation of signals from single markers caused by population stratification were examined by Q-Q plot and a genomic control was applied when necessary. In addition to these single marker-based analyses, we performed haplotype-based analyses to investigate the combinatorial effects of multiple SNPs. The genetic association software Plink was utilized for logistic model regressions23. Haploview was used to construct linkage disequilibrium (LD) structures and haplotype-based analyses24. A regional association plot was created by LocusZoom25. In general, Bonferroni corrections were applied for multiple comparisons. For single marker-based analyses, the threshold of P values was 0.05/15 = 0.003.
Bioinformatics analyses
Two bioinformatics tools were utilized in this study. SIFT26 was used to evaluate the potential biological significance for targeted SNPs. In addition, the effects of targeted SNPs on gene expressions from multiple normal human tissues were examined using the GTEx database27. Relevant plots were made using the R project ggplot package28.
Results
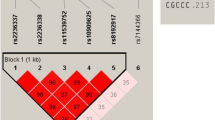
A missense SNP, rs8192917 (Arg55Gln), was identified to be significantly associated with status of vitiligo in our study subjects (Fig. 1). The C allele of this SNP increased the risk of vitiligo by approximately 40% (OR = 1.39, P = 1.92 × 10−8, Table 2). The significant association signals of this SNP were identified in all three genetic modes, although the additive mode seemed to be most powerful. No other SNP showed significance in single marker-based association analyses. The LD structures constructed using data from the 15 genotyped SNPs are shown in Supplemental Fig. S1. Two 2-SNP LD blocks, including rs2236337-rs2236338, rs6573910-rs6573911, were identified, and no significant LD blocks were found in the haplotype-based analyses (Supplemental Table S1). The Q-Q plot was made based on the results of single marker-based association (Supplemental Fig. S2). No significant inflations of association signals can be identified from this plot.

Regional association plot of 15 genotyped SNPs with vitiligo. The most significant SNP (rs8192917) was used as reference to calculate the r2.
The eQTL data for rs8192917 extracted from GTEx showed that this SNP was significantly associated with gene expression of GZMB in human tibial nerve tissue (P = 0.000074, Effect size = 0.28, Supplemental Fig. S3). The result of biological function analyses on rs8192917 using SIFT was “tolerated”, which indicated that this missense SNP would still have a very limited impact on a protein with this mutation.
Discussion
In this study, we evaluated the genetic association between 15 polymorphisms of GZMB and diagnosis with vitiligo based on 3,120 study subjects with Chinese Han ancestry. The results of our single marker-based analyses showed that the C allele of rs8192917indicates an approximately 40% increase in the risk of developing vitiligo in a Chinese population. Compared to an OR of 1.28, as reported by Jin et al. in their GWAS meta analyses on European populations16, our result was slightly higher, at 1.39. This difference may be due to the different ethnicities of the study subjects. The direct effect of this SNP in both studies was the same. In the European populations, researchers have identified a very high LD pattern among the three non-synonymous SNPs (rs8192917, rs11539752 and rs2236338), resulting in alternative protein haplotypes QPY/RAH29. Considering that it is insufficient to draw a reliable conclusion from some SNPs analyses30,31,32, we conducted haplotype analyses and identified a clue for this LD pattern among the three SNPs. However, the LD among these SNPs were not as strong as identified from Europeans. This difference might be due to the difference in population background.
There are several limitations in this study. First, we included only SNPs located within the GZMB gene region. However, for most complex disorders, gene expression are often affected by variations located in upstream or downstream regulatory regions (±30 kb) of the targeted gene. Second, the length of GZMB is approximately 3,000 bp. Based on data from the 1000 genome project, a rough estimation of the genetic variations in this gene is approximately 300. It is thus impossible to capture all the genetic information of GZMB. Furthermore, in order to restrict population stratification we have recruited samples by restricting the subjects with a stable living region33,34, but the potential population stratification could not be excluded completely. Therefore, in future studies, DNA sequencing of the upstream and downstream regulatory regions of GZMB will be necessary to fully evaluate the genetic contributions to the risk of vitiligo.
In summary, we conducted a candidate gene-based association study to investigate the potential genetic contributions of GZMB to the susceptibility of vitiligo. The association signal was identified by single marker-based analyses for a non-synonymous coding SNP rs8192917. Our positive results provide additional supportive evidence that GZMB gene is an important locus for vitiligo in Han Chinese population, and are useful for informative assessment of genetic risk for vitiligo in Han Chinese individuals. Given of unknown complex mechanisms in the etiology of vitiligo, followed-up sequencing-based research would be desired in the future to investigate the genetic architecture of the genomic region of GZMB and its relationship with vitiligo-related phenotypes.
References
Ezzedine, K., Eleftheriadou, V., Whitton, M. & Geel, N. V. Vitiligo. Lancet 386, 74–84 (2015).
Kruger, C. & Schallreuter, K. U. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. International journal of dermatology 51, 1206–1212, https://doi.org/10.1111/j.1365-4632.2011.05377.x (2012).
Majumder, P. P., Das, S. K. & Li, C. C. A genetical model for vitiligo. American journal of human genetics 43, 119–125 (1988).
Bhatia, P. S. et al. Genetic nature of vitiligo. Journal of dermatological science 4, 180–184 (1992).
Hafez, M., Sharaf, L. & Abd elNabi, S. M. The genetics of vitiligo. Acta dermato-venereologica 63, 249–251 (1983).
Das, S. K., Majumder, P. P., Majumdar, T. K. & Haldar, B. Studies on vitiligo. II. Familial aggregation and genetics. Genetic epidemiology 2, 255–262 (1985).
Guan, F. et al. Association of PDE4B polymorphisms and schizophrenia in Northwestern Han Chinese. Hum Genet. 131, 1047–1056 (2012).
Guan, F. et al. MIR137 gene and target gene CACNA1C of miR-137 contribute to schizophrenia susceptibility in Han Chinese. Schizophr Res. 152, 97–104 (2014).
Chen, G., Guan, F., Lin, H., Li, L. & Fu, D. Genetic analysis of common variants in the HDAC2 gene with schizophrenia susceptibility in Han Chinese. Journal of human genetics. 60, 479–484 (2015).
Guan, F. et al. Evaluation of genetic susceptibility of common variants in CACNA1D with schizophrenia in Han Chinese. Scientific reports. 5, 12935 (2015).
Zhang, B. et al. Common variants in SLC1A2 and schizophrenia: Association and cognitive function in patients with schizophrenia and healthy individuals. Schizophr Res. 169, 128–134 (2015).
Guan, F. et al. Evaluation of association of common variants in HTR1A and HTR5A with schizophrenia and executive function. Scientific reports. 6, 38048 (2016).
Guan, F. et al. Evaluation of voltage-dependent calcium channel γ gene families identified several novel potential susceptible genes to schizophrenia. Scientific reports. 6, 24914 (2016).
Jin, Y. et al. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. The New England journal of medicine 362, 1686–1697, https://doi.org/10.1056/NEJMoa0908547 (2010).
Jin, Y. et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nature genetics 44, 676–680, https://doi.org/10.1038/ng.2272 (2012).
Jin, Y. et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nature genetics 48, 1418–1424, https://doi.org/10.1038/ng.3680 (2016).
Birlea, S. A., Gowan, K., Fain, P. R. & Spritz, R. A. Genome-wide association study of generalized vitiligo in an isolated European founder population identifies SMOC2, in close proximity to IDDM8. The Journal of investigative dermatology 130, 798–803, https://doi.org/10.1038/jid.2009.347 (2010).
Shen, C. et al. Genetic Susceptibility to Vitiligo: GWAS Approaches for Identifying Vitiligo Susceptibility Genes and Loci. Frontiers in genetics 7, 3, https://doi.org/10.3389/fgene.2016.00003 (2016).
Spritz, R. A. & Andersen, G. H. Genetics of Vitiligo. Dermatologic clinics 35, 245–255, https://doi.org/10.1016/j.det.2016.11.013 (2017).
Dahl, C. A. et al. Isolation of a cDNA clone encoding a novel form of granzyme B from human NK cells and mapping to chromosome 14. Hum Genet 84, 465–470 (1990).
Crosby, J. L., Bleackley, R. C. & Nadeau, J. H. A complex of serine protease genes expressed preferentially in cytotoxic T-lymphocytes is closely linked to the T-cell receptor α- and δ-chain genes on mouse chromosome 14. Genomics 6, 252–259 (1990).
Guan, F. et al. Association study of a new schizophrenia susceptibility locus of 10q24.32-33 in a Han Chinese population. Schizophr Res. 138, 63–68 (2012).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols 4, 1073–1081 (2009).
Lonsdale, J. et al. The Genotype-Tissue Expression (GTEx) project. Nature genetics 13, 307–308 (2013).
Team, C. R. & Team RDC., R. A Language And Environment For Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria. Computing 1, 12–21 (2016).
Ferrara, T. M. et al. Risk of generalized vitiligo is associated with the common 55R-94A-247H variant haplotype of GZMB (encoding granzyme B). J Invest Dermatol. 133(6), 1677–9 (2013).
Guan, F. et al. A population-based association study of 2q32.3 and 8q21.3 loci with schizophrenia in Han Chinese. Journal of psychiatric research. 47, 712–717 (2013).
Yang, H. et al. 4q22.1 contributes to bone mineral density and osteoporosis susceptibility in postmenopausal women of Chinese Han population. PloS one. 8, e80165 (2013).
Guan, F. et al. Two-stage association study to identify the genetic susceptibility of a novel common variant of rs2075290 in ZPR1 to type 2diabetes. Scientific reports. 6, 29586 (2016).
Guan, F. et al. Two-stage replication of previous genome-wide association studies of AS3MT-CNNM2-NT5C2 gene cluster region in a large schizophrenia case-control sample from Han Chinese population. Schizophr Res. 176, 125–130 (2016).
Jia, X. et al. Two-stage additional evidence support association of common variants in the HDAC3 with the increasing risk of schizophrenia susceptibility. American journal of medical genetics. Part B, Neuropsychiatric genetics. 171, 1105–1111 (2016).
Author information
Authors and Affiliations
Contributions
Authors Xu M.F. and Xiao S.X. conceived and designed the study. Xu M.F. and Chen G. carried out candidate SNPs selection and statistical analyses. Xu M.F., Liu Y., Liu Y.L. and Li X.L. conducted subject screening. Xu M.F., Liu Y., Liu Y.L., Li X.L. and Dong W. contributed to the collection and preparation of control DNA samples. Xu M.F. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, M., Liu, Y., Liu, Y. et al. Genetic polymorphisms of GZMB and vitiligo: A genetic association study based on Chinese Han population. Sci Rep 8, 13001 (2018). https://doi.org/10.1038/s41598-018-31233-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-31233-8
This article is cited by
-
Exploring Therapeutic Approaches for Vitiligo: An Inclusive Review from Translational Modalities to Alternative Therapies
Current Dermatology Reports (2024)
-
Association of GZMB polymorphisms and susceptibility to non-segmental vitiligo in a Korean population
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
