
Abstract
Brugada syndrome (BrS) is an inherited cardiac arrhythmia commonly associated with SCN5A mutations, yet its ionic mechanisms remain unclear due to a lack of cellular models. Here, we used human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) from a BrS patient (BrS1) to evaluate the roles of Na+ currents (INa) and transient outward K+ currents (Ito) in BrS induced action potential (AP) changes. To understand the role of these current changes in repolarization we employed dynamic clamp to “electronically express” IK1 and restore normal resting membrane potentials and allow normal recovery of the inactivating currents, INa, ICa and Ito. HiPSC-CMs were generated from BrS1 with a compound SCN5A mutation (p. A226V & p. R1629X) and a healthy sibling control (CON1). Genome edited hiPSC-CMs (BrS2) with a milder p. T1620M mutation and a commercial control (CON2) were also studied. CON1, CON2 and BrS2, had unaltered peak INa amplitudes, and normal APs whereas BrS1, with over 75% loss of INa, displayed a loss-of-INa basal AP morphology (at 1.0 Hz) manifested by a reduced maximum upstroke velocity (by ~80%, p < 0.001) and AP amplitude (p < 0.001), and an increased phase-1 repolarization pro-arrhythmic AP morphology (at 0.1 Hz) in ~25% of cells characterized by marked APD shortening (~65% shortening, p < 0.001). Moreover, Ito densities of BrS1 and CON1 were comparable and increased from 1.0 Hz to 0.1 Hz by ~ 100%. These data indicate that a repolarization deficit could be a mechanism underlying BrS.
Similar content being viewed by others


Introduction
The Brugada syndrome (BrS) is a rare cardiac rhythm disorder associated with an increased risk of malignant ventricular arrhythmias1,2. The signature ‘coved-type’ ST-segment elevation in the right precordial leads (V1–V3) of the electrocardiogram (ECG) may occur spontaneously or be induced by a provocative drug test with sodium channel blocking drugs1,2. Fetal arrhythmic events in BrS often occur during sleep/rest, a condition associated with slow heart rates3.
SCN5A encodes the pore-forming subunit (Nav1.5) of the cardiac sodium channels. SCN5A variants account for ~80% of BrS, with known genetic mutations4, and there are over 300 genetic variants in SCN5A associated with BrS4,5. Several studies have established loss of sodium channel function associated with these SCN5A variants5,6. However, the low penetrance and variable expressivity of SCN5A reported for BrS raise questions about the quantitative role of the Na+ current in the ionic mechanism of BrS.
A repolarization disorder hypothesis of BrS has been made based on studies using perfused ventricular wedges and isolated cardiac myocytes of animals7,8. Yet the role of the decreased Nav1.5 current (INa) remains to be defined. The genetically engineered haploinsufficient (SCN5A+/−)9 and SCN5A1798insD/+10 mouse models, and the SCN5AE558X/+ pig model11, have shown mainly conduction defects without the loss-of-dome like increased phase-1 repolarization change in the action potential (AP). Moreover, from the two latest reports in which BrS patient-specific human induced pluripotent stem cells (hiPSC) derived cardiomyocytes (hiPSC-CMs) were adopted, no loss-of-dome like AP change was observed12,13.
In the current study, we selected hiPSC-CMs generated from a BrS patient with a severe reduction in INa14. Next, we converted the hiPSC-CMs to more mature, native ventricular myocyte-like cells by in silico injection of a synthetic inward rectifier K+ current (IK1)15 and challenged the cells with slow pacing frequencies. We then identified an INa deficiency-dependent loss-of-INa basal AP pattern of BrS at normal heart rates and an increased phase-1 repolarization proarrhythmic AP pattern at a low heart rate. We further obtained evidence that associated the heart rate-induced AP changes with transient outward K+ current (Ito). We therefore conclude that a loss-of-INa and an elevated Ito could together make the ventricular BrS cardiomyocytes undergo proarrhythmic changes.
Results
Impact of SCN5A mutations of BrS1 on SCN5A/Nav1.5 expression and I Na
A BrS patient with p.A226V and p.R1629X mutations of SCN5A was selected in this study (Fig. 1). First, Tsa201 cells heterologously expressing WT-, A226V- and R1629X-SCN5A were prepared. Immunofluorescence (Fig. 2Aa) and Western blotting (Fig. 2Ab) assays showed that, compared with WT control, A226V-SCN5A cells expressed comparable levels of full-length Nav1.5, whereas R1629X-SCN5A cells showed a much lower level and smaller size of a truncated Nav1.5. Cells co-expressing A226V-SCN5A and R1629X-SCN5A showed both the full length and truncated Nav1.5. Next, INa was recorded in tsa201 cells heterologously expressing WT-, A226V- and R1629X-SCN5A. The average INa density (pA/pF) in A226V-SCN5A cells was ~50% of WT-SCN5A (p < 0.01) and it was almost undetectable in R1629X-SCN5A cells (Table S1, Fig. 2B). The steady state (SS)-inactivation (Fig. 2C) and -activation (Fig. 2D) of INa in A226V-SCN5A cells were largely unaffected (Table S1).

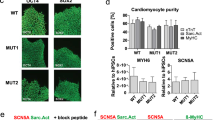
Characterization of hiPSC line and hiPSC-CMs. Representative data from BrS hiPSCs are presented. (A) The expression of pluripotent stem cell markers Oct-4, SSEA-4, Tra-1–60 and Tra-1–81; (B) Teratoma formation in SCID mice after hiPSCs injection confirmed by identifying three primitive germ layers: intestine (top) for endoderm, cartilage (middle) for mesoderm and neuroepithelium (bottom) for ectoderm. (C) Normal karyotype identified with BrS hiPSCs. (D) markers of cardiomyocytes identified with hiPSC-CMs. Top: α-actinin; Middle: β-MHC; and Bottom: cardiac titin.

Expression of SCN5A/Nav1.5 and INa density documented in tsa201 heterologous expression system. tsa201 cells transfected with equal amount of WT-SCN5A, A226V-SCN5A, R1629X-SCN5A and A226V-SCN5A + R1629X-SCN5A (50% each) were assayed for SCN5A/Nav1.5 expression and INa density. (Aa) Representative immunofluorescence staining images of Nav1.5 (purple) in tsa201cells. Arrows indicate the Nav1.5 staining. Levels of Nav1.5 were semi-quantified and plotted in a bar-graph (n = 3). (Ab) Representative Western blotting image of Nav1.5 in tsa201 transfected cells (n = 3). (B) Peak INa density measured in transfected tsa201 cells. (C) and (D) Steady state (SS)-inactivation and activation curves of INa measured in tsa201 expressing WT- and A226V-SCN5A. *p < 0.05; vs. WT-SCN5A.
Next, a ~50% reduction of the SCN5A mRNA was noted in hiPSC-CMs generated from the BrS patient (BrS1) compared with the sibling control (CON1) indicating a possible nonsense-mediated decay (Fig. 3A). The INa density measured in BrS1 was ~25% of CON1 (Fig. 3B,C). Relative to the CON1, BrS1 showed marginal/moderate changes in the rate of SS-inactivation and activation, and a more significant change in the rate of recovery from inactivation (Table S1, Fig. 3D–F).

SCN5A and Nav1.5 expressions and INa measured in hiPSC-CMs. (A) Level of SCN5A mRNA in BrS1 (hiPSC-CMs from a BrS patient with compound SCN5A mutations) and CON1 (the sib of BrS1) determined by qPCR. n = 3. (B) Representative traces of sodium currents in the control (CON1) and patient (BrS1). (C) Peak INa density measured in BrS1 and CON1. (D–F) Steady state (SS)-inactivation, activation and recovery from inactivation curves of INa. Values given are mean ± SEM. †p < 0.01; vs. CON1.
Identification of a loss-of-I Na basal AP pattern with BrS1
HiPSC-CMs are known for their lack of IK1, which is responsible for their depolarized resting membrane potential (RMP) of ~−60 mV compared with the ~−90 mV of quiescent adult ventricular and atrial myocytes15,16. The more positive resting membrane potential in hiPSC-CMs importantly results in inactivation of the fast INa, leaving very limited INa available during the phase-0 depolarization and thereby potentially masking the impact of INa deficiency on the AP. The depolarized state of hiPSC-CMs could also lead to Ito inactivation and the absence of phase-1 repolarization in hiPSC-CMs16. Indeed, APs recorded from spontaneously contracting CON1 and BrS1, or BrS1 paced at 1.5, 1.0, 0.5, 0.2 Hz, were comparable without displaying a phase-1 repolarization morphology (Table S2, Figure S3).
To overcome this limitation, a synthetic, cardiomyocyte membrane potential-dependent in silico IK1 was injected into hiPSC-CMs using the dynamic clamp technique15 to produce a physiologically polarized resting membrane potential (−84 mV). Thereafter, ventricular-like hiPSC-CMs (IK1positive) from CON1 (Fig. 4A) paced at a 1.0 Hz displayed a phase-1 repolarization AP morphology that resembles normal human ventricular myocytes. In contrast, an AP rounded pattern, clearly distinguishable from the phase-1 repolarization pattern of CON1, was observed in ventricular-like BrS1 (Fig. 4B). Such APs were characterized by over 75% reduction in the maximum upstroke velocity (dV/dtMax) and reduced AP amplitude (APA) and overshoot (Table S3, Fig. 4C). This AP pattern, referred as a “loss-of-INa” AP morphology, reflects a marked INa deficiency in BrS1 during depolarization.

A basal action potential pattern of BrS identified in BrS1. (A and B) Representative AP waveforms recorded from a CON1 and a BrS1 paced at 1.0 Hz. The solid and dashed arrows indicate APA measured at phase-0 and phase-2, respectively. (C) The main AP parameters of CON1 and BrS1 are plotted and compared in bar-graphs. Values given are mean ± SEM. ||p < 0.00001; vs. CON1 (by unpaired t-test).
Identification of a heart rate-induced increased phase-1 repolarization proarrhythmic AP change in BrS1
In BrS patients, sleep and rest are the most common triggers of ST-segment elevation, ventricular arrhythmia, and sudden death1,3. To test the effect of heart rate on the pathophysiology of BrS, APs were recorded from CON1 and BrS1 paced at 1.0 Hz, 0.5 Hz and 0.1 Hz. Responding to reduced pacing frequencies from 1.0 Hz to 0.1 Hz, CON1 showed slight changes in APD (±27% change in APD90 and the maximal shortening per cell was <33%) (Table S4, Fig. 5A, Table S5). In contrast, diverse changes in APD were observed with BrS1 paced at 0.1 Hz. The majority (75%) of BrS1 (the non-early repolarization subgroup) demonstrated a marked prolongation of APD90 or moderate shortening (<28% per cell) like CON1 (Fig. 5B1). However, a fraction (25%) of BrS1 (the early repolarization subgroup) displayed a drastic shortening of APDs (APD90 reduced by 66% in average and ≥50% per cell) accompanied by decreased dV/dtMax and APA, and a more depolarized resting membrane potential (Table S4, Fig. 5B2, Table S5). The marked reduction in APD, particularly the phase-2 APD indicated by an over 50% reduction in the ratio of APD20/APD90, represented an increased phase-1 repolarization AP pattern highly resembles the proarrhythmic loss-of-dome AP change recorded from canine right ventricle epicardium model under the repolarization disorder hypothesis7,8.

Effects of low heart rates on APs of BrS1 and CON1 hiPSC-CMs. (A, B1 and B2) (Top): representative superimposed AP waveforms recorded from CON1 and two BrS1 subgroups (non-early repolarization and early repolarization), sequentially paced at 1.0 Hz, 0.5 Hz and 0.1 Hz. (A, B1 and B2) (Bottom): superimposed AP waveforms continually recorded from CON1 and two BrS1 subgroups sequentially paced at 1.0 Hz and 0.1 Hz. (C) Main AP parameters plotted and compared among groups in bar-graphs. Values given are mean ± SEM. Statistics were performed with two-way repeated measures ANOVA followed by the Bonferroni post hoc testing. *p < 0.05; †p < 0.01; ‡P < 0.001. Black symbols: Intra-(sub) group comparisons. 0.1 Hz/0.5 Hz vs. 1.0 Hz. Pink symbols: vs. BrS1 (non-ER subgroup) at the same frequency. Green symbols: vs. CON1 subgroup at 1.0, 0.5 and 0.1 Hz.
On the other hand, CON2 and BrS2 (at both 24 °C and 34 °C) paced at 1.0 and 0.1 Hz displayed APDs within the normal range, similar to CON1. In agreement with that observed in tsa201 cells17, we observed an increased dV/dtMax and APA/overshoot in hiPSC-CMs at 34 °C compared with that 24 °C, indicating an increased INa at 34 °C (Fig. 6). Normal levels of INa were observed with BrS2 and dV/dtMax at 34 °C and 24 °C were all ≥190, which is greater than the 110~185 level of controls (CON1 and CON2) and significantly larger than the 41.9 ± 5.3 levels observed in BrS1.

Validation of the rate-dependent effects on APs in BrS2 and CON2. (A) Representative superimposed AP waveforms of a CON2 cell paced at 1.0 and 0.1 Hz at 24 °C. (B and C) Representative superimposed AP waveforms of BrS2 cells paced at 1 and 0.1 Hz at 24 °C and 34 °C, respectively. (D) Main AP parameters are plotted and compared among groups in bar-graphs. Values given are mean ± SEM. Statistics were performed with two-way repeated measures ANOVA followed by the Bonferroni post hoc testing (by paired t-test). *p < 0.05; ‡P < 0.001.
Evaluating the role of I to in mediating the heart rate-dependent AP changes
In cardiac myocytes, Ca2+-independent Ito consists of a fast recovery component (Ito,f) mediated mainly by Kv4.3 subunits and a slow recovery component (Ito,s), mediated by Kv1.4 subunits. Ito contributes to the phase-1 repolarization of the AP. Slow heart rates favor the recovery of Ito18,19, whereas increased Ito levels could accentuate the phase-1 notch of the AP20,21,22.
To explore the potential involvement of Ito in heart rate-induced AP changes, the level of Ito in CON1 and BrS1 paced at 1.0 Hz and 0.1 Hz were measured and compared (Fig. 7). Ito densities of both CON1 and BrS1 paced at 1.0 Hz were comparable (~5 pA/pF at 40 mV), which is ~50% of that in isolated human sub-epicardial ventricular myocytes (10.6 ± 1.08 pA/pF at 40 mV)18. A ~100% increase (p < 0.00001) of Ito densities was recorded in both CON1 and BrS1 when the heart rate was reduced to 0.1 Hz. Moreover, variations in the levels of Ito among different hiPSC-CMs, particularly of those paced at 0.1 Hz, were identified in a fraction of cells (19~23%) which showed much higher Ito densities. Our data suggest potential heterogenic expression of Ito among hiPSC-CMs and Ito,s could be the dominant component of Ito.

Effects of heart rates on Ito in hiPSC-CMs. (A-left and B-left) Representative traces of Ito currents recorded from CON1 (n = 26) and BrS (n = 22) paced at 1.0 Hz and 0.1 Hz. (A-middle and B-middle) I-V relationships of Ito show the current densities. (A-right and B-right) Graphic demonstrations and comparisons of Ito (recorded at +50 mV) in each cell are presented. Values given are mean ± SEM. §p < 0.0001; ||p < 0.00001; vs. 1.0 Hz (by paired t-test and one-way ANOVA test). ‡P < 0.001, vs. low Ito group.
To further validate the role of Ito in heart rate-induced AP changes, BrS1 that displayed an increased phase-1 repolarization AP change at 0.1 Hz were treated with 4-Aminopyridine (4-AP), which is known to block both Ito,f and Ito,s23,24 at higher concentrations (0.5 mM to 10 mM could effectively block 55−85% of Ito)22. As 4-AP at 1.0 mM and above could also block the rapid and slow components of the delayed rectifier K+ currents (IKr and IKs) by over 25% and 39%, respectively25, we applied 0.05 mM and 0.5 mM 4-AP to BrS1 and noted that 0.05 mM 4-AP caused moderate APD prolongations, while 0.5 mM 4-AP completely reversed the increased phase-1 repolarization (Table S6, Fig. 8A). The significant increase in the ratio of APD20/APD90 suggested that 4-AP treatment recovered the loss of phase-2 repolarization.

Effects of 4-AP and flecainide on APs. (A) Representative superimposed AP waveforms of a BrS1 cell paced at 0.1 Hz and subsequently exposed to 0.05 mM and 0.5 mM 4-AP. Main AP parameters (showing only the results of 0.5 mM 4-AP) are presented in bar-graphs. (B) Representative superimposed AP waveforms of a V-like BrS1 hiPSC-CM paced at 1.0 Hz followed by flecainide (10 µM) treatment. Main AP parameters are presented in bar-graphs. Values given are mean ± SEM. *p < 0.05; †p < 0.01; §p < 0.0001; ||p < 0.00001 (by paired t-test); vs. 0.1 Hz (for 4-AP) or 0.1 Hz (for flecainide).
Blocking I Na induced increased phase-1 repolarization-like change in BrS1
Ajmaline and flecainide are commonly employed in the clinical setting as challenges to attempt to unmask the characteristic BrS ECG phenotype and its associated arrhythmic events7. We observed that BrS1 paced at 1.0 Hz responded to flecainide (10 µM) with a significant shortening in APDs (with an average of 43.6% reduction in APD90) accompanied by reductions in dV/dtMax from 31.4 ± 7.7 to 12.6 ± 2.7 V/s (p < 0.01) and APA/overshoot, confirming an INa inhibition (Table S7 and Fig. 8B). Control cardiomyocytes (CON2) response to flecainide with reduced action potential amplitude/overshoot and dV/dtMax similar to BrS1, indicating a spike and dome morphology change.
Computer simulation of the effects of I to and I Ca,L on APDs
Adopting the O’Hara-Rudy model (2013) of human epicardial ventricular myocytes26, computer simulation was performed to examine the effects of Ito and ICa,L on APDs. While APD shortening was not seen in the presence of lower INa (INa reduced to 15%), it was observed with INa reduced to 15% (no effect with 20%) and Ito increased to 5 folds (Figure S5A). On the other hand, the shortened APD was not observed with reduced ICa,L in the presence of reduced INa (Figure S5B).
Discussion
In cardiomyocytes derived from a BrS patient (BrS1) with a severe SCN5A haploinsufficiency, and an over 75% reduction in INa,, we identified a highly distinguishable loss-of-INa AP pattern at normal pacing frequency (1.0 Hz) and an increased phase-1 repolarization AP pattern characterized by marked reduction in APDs and more depolarized resting membrane potential at a slow pacing frequency (0.1 Hz). Observation of these changes in repolarization required use of dynamic clamp to “electronically” express IK1 and allow recovery from inactivation of the major inactivating currrents INa, ICa and Ito. In contrast, cardiomyocytes without a marked INa reduction (BrS2) and two normal controls (CON1 and CON2) failed to show heart rate-induced AP abnormalities. Importantly, similar low levels INa were observed in four separate hiPSC lines from the same patient (Table S1), and one line was selected for our experiments. We further correlated the reduced heart rate (from 1.0 Hz to 0.1 Hz) with a ~100% increase in Ito, suggesting that a higher Ito acts together with a low INa contributes to the APD shortening phenotype in BrS1.
Thus, the results from the current study suggest repolarization abnormalities. The repolarization hypothesis of BrS implies that the loss-of-dome change is limited to epicardial myocytes and corresponds to a transmural dispersion of repolarization causing an ST-segment elevation in the ECG7. The fact that cardiac myocytes from a BrS patient predisposed with a severe INa deficiency (such as BrS1) could develop arrhythmic changes at slow heart rates, and also associated with an elevated Ito, supports the repolarization disorder hypothesis that a low INa in conjunction with a high Ito could result in an increased phase-1 repolarization7,8.
Interestingly, the heart rate-sensitive and Ito-mediated basal and pro-arrhythmogenic AP phenotypes identified in BrS hiPSC-CMs might mirror a switch between the daytime/active/asymptomatic and sleep/bradycardia/disease onset states seen in many BrS patients. Even with a severe INa deficiency, APs of BrS1 remain largely ‘normal’ at normal heart rates which keep patients in ‘event-free’ clinical status. A precipitating factor is needed, such as sodium channel blockade7, to trigger or unmask arrhythmic events. Ito, coordinated by INa and ICa,L, plays a key role in shaping the AP of ventricular epicardial myocytes. Ito,f and Ito,s23,24 coexist in ventricular myocytes and are distinguishable by the either tens or thousands of milliseconds taken for recovery from inactivation23. The expressions of Kv1.4 and Kv4.3 genes in human right ventricular myocytes have been confirmed, and the level of Kv1.4 is ~37.5% that of Kv4.3 and the Kv4.3-dependent KChIP2 is >7-fold that of Kv4.327. Ito,s plays a role in ventricular myocytes and slow heart rates could facilitate its recovery from inactivation.. An accentuated phase-1 notch in canine epicardial ventricular myocytes paced at 0.125 Hz 19and 0.5 Hz20, and in ferret ventricle myocytes paced at 0.2 Hz23, was noted. Moreover, canine ventricular myocytes paced at 0.125 Hz were more vulnerable to an ischemia-induced loss-of-dome AP change28. Furthermore, it has been shown that hiPSC-CMs responded to 1.0 Hz, 0.5 Hz and 0.1 Hz with 20%, 40% and 100% recovery of Ito, respectively; and hiPSC-CMs express a relatively high level of Kv1.4 (~80% of Kv4.3) and a much lower level of KChIP2 compared Kv4.328. Our data from hiPSC-CMs are consistent with that of Cordeiro et al.28. and indicate that Ito,s could be a main component of Ito in hiPSC-CMs and play a dominant role at 0.1 Hz when there is more time to recover from inactivation. Our data further suggest that, like that among different subtypes of human ventricular myocytes such as those from the right ventricle pericardium and endocardium, Ito could be heterogeneously expressed among hiPSC-CMs and a small fraction of cells that express a distinguishable higher level of Ito could behave like the right ventricle epicardium myocytes prone to the ‘loss-of-dome’ AP change (Fig. 7). Treatment with 4-AP also reversed the increased phase-1 repolarization which we attribute to block of Ito channels (rather than potentiation of ICa,L which was reported in heterologous experiments29.
Our experimental findings from hiPSC-CMs are supported by results from a computer simulation study using the O’Hara-Rudy non-diseased human ventricular epicardial myocyte model (2011)26. When INa was reduced to 10~30% of the control level, a subsequent 1.7~2.4-fold Ito increments was associated with ‘loss-of-dome’ AP change30. Ca2+ handling was not investigated in our study, but has been reported to have a role in arrhythmogenesis in an hiPSC-CM model of Brugada13.
Although BrS patient-derived hiPSC-CMs represent the most advanced model that reflects the basal cellular phenotype associated with SCN5A defects and INa deficiency, further dissecting the mechanism behind the BrS is met with limitations. Beside a weaker than normal inward rectifier current (IK1) even when IK1 is increased using dynamic clamp, these cells have lower expression of slowly reactivating Ito. Thus, hiPSC-CMs do not fully recapitulate the epicardial action potential phenotype, and the spike and dome configuration of the AP is not obvious.
Despite our novel and compelling findings, our study has limitations. First, our data may not fully explain the mechanisms behind many BrS cases in which the SCN5A defects are associated with only a milder/moderate INa deficiency. Indeed, our data from BrS2 demonstrated that the heterologous T1620M variant5,17 shows a minimal, if any, impact on INa in hiPSC-CMs recorded at both 24 °C and 34 °C, reflected by the normal dV/dtMax and APA/overshoot. A recent report further supports this notion by showing normal APs from the hiPSC-CMs of BrS patients without any genetic mutations12. The likely different expression profiles of Ito,f and Ito,s in hiPSC-CMs (Ito,s –dominant, demonstrated in this study and a previous report28) compared with that in human epicardial ventricular myocytes (Ito,f –dominant23,24) may compromise the value of this model. This may also explain the lack of consistency between the cellular phenotypes and the clinical findings of the patients. Second, we acknowledge that we did not correct these mutations with gene editing in the hiPSC-CMs to rescue the disease phenotype. However, we believe that using a healthy sibling and a standard cell line are appropriate controls in this study, as done previously by our lab and others31,32,33,34,35. Finally, although hiPSC-CMs were studied a longer than 30 days after differentiation, prolonged culture times can have effects on INa properties36. These effects should be taken into account when interpreting the data and further studies are needed to access the potential impact on hiPSC-CMs as an electrophysiological model.
Conclusions
Using hiPSC-derived cardiomyocytes from a BrS patient with SCN5A mutations, we demonstrate that a severe INa deficiency could lead to a remodeled baseline APs vulnerable to heart rate-induced, Ito-sensitive proarrhythmic increased phase-1 repolarization changes. Our data support a coordinated role of INa and Ito in the mechanism of BrS.
Materials and Methods
For a full description of the materials and methods, see Supplemental Material.
hiPSC-CMs from BrS patients and controls
To obtain a BrS hiPSC-CM model with marked INa deficiency, a BrS patient (II-2) with compound heterozygous mutation in SCN5A (a missense mutation: c.677 C > T, p.A226V, and a nonsense mutation: c.4885 C > T, p.R1629X)14, and a sibling control, were included in this study (Figure S1). All experimental protocols were approved by the institutional review board (Institutional Review Board of SingHealth, Singapore) and all methods were performed following the relevant guidelines and regulations. Clinical data and biological samples were taken following written informed consent. hiPSCs were generated from the dermal fibroblasts of the BrS patient and sibling control (Fig. 1). All hiPSC lines were expanded as adherent cultures in feeder-free conditions on Matrigel-coated dishes in the presence of chemically defined medium (E8 Essential Medium, Life Technologies). Differentiation of hiPSC to cardiomyocytes (CMs) was performed following a previously reported protocol based on small molecules-mediated canonical Wnt pathway modulation37. Four hiPSC clones, which showed good self-renewal potential and cardiac differentiation efficiency, were selected. Eventually, single CON1 and BrS1 clone, evidenced by yielding >55% cardiac troponin T (cTnT) positive hiPSC-CMs quantified by fluorescence-activated cell sorting (FACS) analysis (Figure S6), were adopted. HiPSC-CMs were further stained with anti-α-actinin, anti-β-MHC and anti-cardiac titin antibodies (Fig. 1D). The BrS1 and CON1 hiPSC-CMs were dissociated into single cells 35–45 days after cardiac differentiation and used for electrophysiology assays.
We also used cardiomyocytes (CON2), a commonly used control hiPSC line (iCell® Cardiomyocytes, from a female Caucasian with the age unknown), and a genome edited BrS hiPSC-CM line (BrS2) which carries the p. T1620M SCN5A mutation known to have a milder impact on INa in heterologous expression systems4,6,17. Both lines were commercially obtained from Cell Dynamic International (Madison, WI, USA).
Whole-cell patch-clamp assays
An Axopatch 200B Amplifier controlled by pClamp10 software together with a Digidata 1440 acquisition system (Molecular Devices, Sunnyvale, USA) was used to record ionic currents and APs.
INa in tsa201 cells and hiPSC-CMs32, Ito12,24,28 and the ultra-rapid delayed rectifier K+ current (IKur)38,39 in hiPSC-CMs were measured using standard voltage-clamp protocols.
Dynamic clamp was performed using a Cybercyte System from Cytocybernetics (Buffalo, USA)15. With an injection of a synthetic IK1 current, the ventricular-like hiPSC-CMs (IK1positive) were defined as having AP amplitude (APA) over 100 mV, APD at 90% repolarization (APD90) over 200 ms and lack of IKur38, which is highly expressed in the atrial-like human embryonic stem cell-derived cardiomyocytes34 and hiPSC-CMs (Figure S2).
Statistical analyses
Numerical data are presented as mean ± standard deviation (SD) or mean ± standard error of the mean (SEM). Comparisons were made with unpaired and paired (two-tailed) Student’s t-test, one-way repeated measures ANOVA followed by the Tukey’s post hoc testing, and two-way repeated measures ANOVA followed by the Bonferroni post hoc testing using GraphPad Prism 5.0 (GraphPad Software, La Jolla, USA). A p-value of <0.05 was considered statistically significant.
References
Brugada, P. & Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. Journal of the American College of Cardiology 20, 1391–1396 (1992).
Priori, S. G. et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart rhythm: the official journal of the Heart RhythmSociety 10, 1932–1963, https://doi.org/10.1016/j.hrthm.2013.05.014 (2013).
Nademanee, K. et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation 96, 2595–2600 (1997).
Kapplinger, J. D. et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart rhythm: the official journal of the Heart Rhythm. Society 7, 33–46, https://doi.org/10.1016/j.hrthm.2009.09.069 (2010).
Priori, S. G. et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation 102, 2509–2515 (2000).
Chen, Q. et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296, https://doi.org/10.1038/32675 (1998).
Yan, G. X. & Antzelevitch, C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation 100, 1660–1666 (1999).
Aiba, T. et al. Cellular basis for trigger and maintenance of ventricular fibrillation in the Brugada syndrome model: high-resolution optical mapping study. J Am Coll Cardiol 47, 2074–2085, https://doi.org/10.1016/j.jacc.2005.12.064 (2006).
Papadatos, G. A. et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proceedings of the National Academy of Sciences of the United States of America 99, 6210–6215, https://doi.org/10.1073/pnas.082121299 (2002).
Remme, C. A. et al. Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation 114, 2584–2594, https://doi.org/10.1161/CIRCULATIONAHA.106.653949 (2006).
Park, D. S. et al. Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. The Journal of clinical investigation 125, 403–412, https://doi.org/10.1172/JCI76919 (2015).
Veerman, C. C. et al. hiPSC-derived cardiomyocytes from Brugada Syndrome patients without identified mutations do not exhibit clear cellular electrophysiological abnormalities. Scientific reports 6, 30967, https://doi.org/10.1038/srep30967 (2016).
Liang, P. et al. Patient-Specific and Genome-Edited Induced Pluripotent Stem Cell-Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Brugada Syndrome. J Am Coll Cardiol 68, 2086–2096, https://doi.org/10.1016/j.jacc.2016.07.779 (2016).
Tan, B. Y. et al. A Brugada syndrome proband with compound heterozygote SCN5A mutations identified from a Chinese family in Singapore. Europace: European pacing, arrhythmias, and cardiac electrophysiology: journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology https://doi.org/10.1093/europace/euv058 (2015).
Bett, G. C. et al. Electronic “expression” of the inward rectifier in cardiocytes derived from human-induced pluripotent stem cells. Heart rhythm: the official journal of the Heart Rhythm. Society 10, 1903–1910, https://doi.org/10.1016/j.hrthm.2013.09.061 (2013).
Meijer van Putten, R. M. et al. Ion channelopathies in human induced pluripotent stem cell derived cardiomyocytes: a dynamic clamp study with virtual IK1. Frontiers in physiology 6, 7, https://doi.org/10.3389/fphys.2015.00007 (2015).
Dumaine, R. et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res 85, 803–809 (1999).
Nabauer, M., Beuckelmann, D. J., Uberfuhr, P. & Steinbeck, G. Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation 93, 168–177 (1996).
Xiao, L. et al. Mechanisms underlying rate-dependent remodeling of transient outward potassium current in canine ventricular myocytes. Circ Res 103, 733–742, https://doi.org/10.1161/CIRCRESAHA.108.171157 (2008).
Liu, D. W., Gintant, G. A. & Antzelevitch, C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ Res 72, 671–687 (1993).
Lukas, A. & Antzelevitch, C. Differences in the electrophysiological response of canine ventricular epicardium and endocardium to ischemia. Role of the transient outward current. Circulation 88, 2903–2915 (1993).
Campbell, D. L., Qu, Y., Rasmusson, R. L. & Strauss, H. C. The calcium-independent transient outward potassium current in isolated ferret right ventricular myocytes. II. Closed state reverse use-dependent block by 4-aminopyridine. The Journal of general physiology 101, 603–626 (1993).
Patel, S. P. & Campbell, D. L. Transient outward potassium current, ‘Ito’, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J Physiol 569, 7–39, https://doi.org/10.1113/jphysiol.2005.086223 (2005).
Amos, G. J. et al. Differences between outward currents of human atrial and subepicardial ventricular myocytes. The Journal of physiology 491(Pt 1), 31–50 (1996).
Arechiga-Figueroa, I. A., Rodriguez-Martinez, M., Albarado, A., Torres-Jacome, J. & Sanchez-Chapula, J. A. Multiple effects of 4-aminopyridine on feline and rabbit sinoatrial node myocytes and multicellular preparations. Pflugers Archiv: European journal of physiology 459, 345–355, https://doi.org/10.1007/s00424-009-0734-3 (2010).
O’Hara, T., Virag, L., Varro, A. & Rudy, Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS computational biology 7, e1002061, https://doi.org/10.1371/journal.pcbi.1002061 (2011).
Gaborit, N. et al. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol 582, 675–693, https://doi.org/10.1113/jphysiol.2006.126714 (2007).
Cordeiro, J. M. et al. Identification and characterization of a transient outward K+ current in human induced pluripotent stem cell-derived cardiomyocytes. Journal of molecular and cellular cardiology 60, 36–46, https://doi.org/10.1016/j.yjmcc.2013.03.014 (2013).
Li, L. et al. Potentiation of high voltage-activated calcium channels by 4-aminopyridine depends on subunit composition. Molecular pharmacology 86, 760–772, https://doi.org/10.1124/mol.114.095505 (2014).
Zhang, J. et al. Cardiac electrophysiological substrate underlying the ECG phenotype and electrogram abnormalities in Brugada syndrome patients. Circulation 131, 1950–1959, https://doi.org/10.1161/CIRCULATIONAHA.114.013698 (2015).
Egashira, T. et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovascular research 95, 419–429, https://doi.org/10.1093/cvr/cvs206 (2012).
Ma, D. et al. Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. International journal of cardiology 168, 5277–5286, https://doi.org/10.1016/j.ijcard.2013.08.015 (2013).
Campbell, K. A., Terzic, A. & Nelson, T. J. Induced pluripotent stem cells for cardiovascular disease: from product-focused disease modeling to process-focused disease discovery. Regenerative medicine 10, 773–783, https://doi.org/10.2217/rme.15.41 (2015).
Terrenoire, C. et al. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. The Journal of general physiology 141, 61–72, https://doi.org/10.1085/jgp.201210899 (2013).
Jouni, M. et al. Toward Personalized Medicine: Using Cardiomyocytes Differentiated From Urine-Derived Pluripotent Stem Cells to Recapitulate Electrophysiological Characteristics of Type 2 Long QT Syndrome. Journal of the American Heart Association 4, e002159, https://doi.org/10.1161/JAHA.115.002159 (2015).
Veerman, C. C. et al. Switch From Fetal to Adult SCN5A Isoform in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Unmasks the Cellular Phenotype of a Conduction Disease-Causing Mutation. Journal of the American Heart Association 6 https://doi.org/10.1161/JAHA.116.005135 (2017).
Lian, X. et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci USA 109, E1848–1857, https://doi.org/10.1073/pnas.1200250109 (2012).
Ravens, U. & Wettwer, E. Ultra-rapid delayed rectifier channels: molecular basis and therapeutic implications. Cardiovascular research 89, 776–785, https://doi.org/10.1093/cvr/cvq398 (2011).
Devalla, H. D. et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO molecular medicine 7, 394–410, https://doi.org/10.15252/emmm.201404757 (2015).
Acknowledgements
We thank Dr. Mark Nowak for technical assistance.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: H.W., R.L., S.A.C. and R.L.R. Performed the experiments: D.M., Z.L., L.J.L., Y.Z., G.L., O.I., J.W., Y.Y.C. and B.K.P. Analyzed the data: H.W., D.M., Z.L., Y.Z. and R.L.R. Contributed reagents/materials/analysis tools: R.L., W.S.T., C.K.C., B.Y.T., D.C., K.L.H., P.L., R.Y.Y.Y., R.L.R., A.D.K. and G.C.L.B. Contributed to the writing of the manuscript: H.W., S.A.C., J.W., C.R.B., A.O.V. and R.L.R.
Corresponding authors
Ethics declarations
Competing Interests
Drs Bett and Rasmusson are co-founders and owners of Cytpocyberentics Inc. which builds and sells the Cybercyte dynamic clamp system used in this study. Cytocybernetics produced Figure S4 on Flecanide block of the action potential. Other authors have no competing financial and non-financial interests as defined by Nature Research, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, D., Liu, Z., Loh, L.J. et al. Identification of an INa-dependent and Ito-mediated proarrhythmic mechanism in cardiomyocytes derived from pluripotent stem cells of a Brugada syndrome patient. Sci Rep 8, 11246 (2018). https://doi.org/10.1038/s41598-018-29574-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29574-5
This article is cited by
-
HiPSC-derived cardiomyocyte to model Brugada syndrome: both asymptomatic and symptomatic mutation carriers reveal increased arrhythmogenicity
BMC Cardiovascular Disorders (2023)
-
Contributions of NaV1.8 and NaV1.9 to excitability in human induced pluripotent stem-cell derived somatosensory neurons
Scientific Reports (2021)
-
Comparing human iPSC-cardiomyocytes versus HEK293T cells unveils disease-causing effects of Brugada mutation A735V of NaV1.5 sodium channels
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
