
Abstract
Members of the freshwater halfbeak genus Dermogenys are hard to identify to the species level, despite several previous attempts to isolate fixed meristic, morphometric and colour pattern differences. This has led to ongoing confusion in scientific literature, records of species occurrence, and entries in museum collections. Here, a DNA barcoding study was conducted on the genus to gain further understanding of its taxonomic status across the Southeast Asian region. Fish were collected from 33 localities, spanning freshwater and brackish habitats in Malaysia, Western Indonesia, Thailand and Vietnam. In total, 290 samples of Dermogenys spp. were amplified for a 651 base pair fragment of the mitochondrial cytochrome oxidase c subunit I (COI) gene. Analysis was able to successfully differentiate the three species: D. collettei, D. siamensis, D. sumatrana; reveal the presence of a new putative species, Dermogenys sp., that was sampled in sympatry with D. collettei at three locations; as well as uncovering two genetic lineages of a fifth species, D. bispina, that display non-overlapping geographical distributions in drainages of northern Borneo; Kudat and Sandakan. This study expands the barcode library for Zenarchopteridae, demonstrates the efficacy of DNA barcoding techniques for differentiating Dermogenys species, and the potential thereof in species discovery.
Similar content being viewed by others

Introduction
The tropical halfbeak genus Dermogenys, commonly known as silver halfbeak, pygmy halfbeak, wrestling halfbeak, Malayan halfbeak, or ‘julong julong’, occurs in the Southeast Asian region. The genus is the smallest in size among zenarchopterids, and inhabits fresh and brackish waters1, being able to tolerate some level of salinity. Halfbeaks are of limited economic value, however they are sometimes encountered in regional fish markets in Cambodia, Thailand and Vietnam.
As a group, Dermogenys has received considerable scientific attention2,3,4,5,6,7,8,9,10,11,12,13,14. The genus is viviparous (bearing live young) and generally limited to freshwater and estuarine habitats, suggesting low dispersal capacity; yet has a wide natural geographical distribution, including large mainland rivers such as the Mekong and drainages of the Indonesian and Philippine archipelagos. These factors mean the genus presents a tractable model for investigation of factors affecting phylogeographical pattern12 in what is a biogeographically complex and biologically diverse region of the tropics15,16.
Historically, Dermogenys taxonomy and systematics have been contentious and challenging. First described in the early 19th Century, in van Hasselt17, the type specimen was referred to as D. pusilla. In the first revision, Mohr18 classified four of the then ten nominal species, considering three to be synonyms of D. pusilla. In a subsequent revision, Brembach19 recognized ten species and three subspecies, with diagnosis based on modified anal fin characters in males. He classified several populations that had consistent differences in the andropodium (male modified anal fin) as D. pusilla. This has led to erroneous classifications throughout museum collections and the literature11. Meisner & Collette1 named a new Dermogenys species from Sabah as D. bispina. This was followed by the discovery of a further four species, differentiated based on phylogenetic analysis of the gonadal histology and embryonic modifications associated with viviparity, and on osteological characters in modified anal fin rays11. Meisner concluded that the osteological characters of anal fin rays in males were diagnostic for species identification. Currently, there are 12 morphologically recognised species of Dermogenys in the Southeast Asian inland waters, as reviewed by Meisner11.
Among the halfbeaks, the genus level diagnostics as described by Rainboth20 are widely used as a guide for field identification. To aid identification at lower taxonomic rank, Meisner & Burns9,10 and Meisner11 developed keys for species level Dermogenys identification based on extensive morphological and morphometric analyses of a large number of anatomical structures. However, taxonomic differentiation among Dermogenys species remains challenging and is difficult in non-specialist facilities due to the small size of the individuals and overlapping morphological characteristics within the genus, requiring a considerable degree of skill and taxonomic expertise. For instance, Meisner11 concluded that the osteological characters are only informative at the generic level, while gonad histology yields characters useful for differentiating species groups within genera, as well as differentiating at genus level between Dermogenys and Nomorhamphus. According to Meisner11, the single diagnostic character for species level identification is the shape of the modified anal fin, however this character is only present in male specimens, and is only discernible through specialised radiography and optimal staining techniques. Although meristic and morphometric characters have proved efficient in resolving taxonomic questions at the species level in many teleost studies21,22,23,24,25,26,27,28,29,30, in the case of Dermogenys, they fail to adequately identify individuals to species level11,19. Likewise, characters like black pigmentation of pelvic and dorsal fins in males and melanophore arrangement from anterior to anal fin in females may aid in sorting the Dermogenys specimens into different groups at the genus level, but not to a precise taxon.
This study therefore attempts to alleviate these taxonomic challenges through a DNA barcoding approach. This technique, which relies on fixed differences in mitochondrial DNA sequences between taxa, has shown to be approximately 90% successful in species identification of freshwater and marine fishes31,32,33,34,35. Once a DNA barcoding database is established, the method offers a rapid way to allocate unknown specimens to correct species names, and can be used to identify individuals that lack defining morphological characters, for example females, larval fishes, or processed fish products36,37,38, among a range of other applications39,40,41,42. The ability for DNA barcoding to identify unknown specimens has proven useful across a wide range of biological disciplines, such as in biosecurity43,44, wildlife forensics45,46,47, phylogenetics31,48,49,50, and more generally in the conservation and management of wildlife51,52,53,54.
To date, the only Dermogenys barcode sequences available in online DNA databases and identified to species level are for D. pusilla, while all other Dermogenys barcodes come from fish that were only identified to genus level. Therefore, this study aims to generate new DNA barcodes for the species obtained during the course of this study, and to assign these to the species level using morphological and molecular tools, providing a robust taxonomic framework for future research on Dermogenys. The findings provide further insights into systematics as well as the phylogenetic relationships of the genus and highlight the complementarity of morphological and molecular characteristics in elucidating the taxonomic status and systematics of this group.
Results
Collection and morphological identification
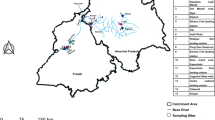
Sample collection across 33 freshwater locations in Thailand, Vietnam, Malaysia and Western Indonesia yielded a total of 290 Dermogenys specimens (Fig. 1, Table 1). Island-wide sampling was conducted in Western Indonesia (Sumatra), however no Dermogenys specimen was obtained from the west coast of this island. Across all locations, most of the individuals collected (70%) were adults and therefore could be putatively assigned to their morphological species using the techniques outlined in the Methods section. This resulted in the identification of five morpho-species: D. collettei Meisner11 (30%), D. sumatrana Bleeker55 (14.8%), D. siamensis Fowler56 (17.6%), D. bispina Meisner & Collette1 (3.1%), and a newly observed group henceforth referred to as Dermogenys sp. (4.8%).

The localities of Dermogenys fish sampled from inland waters of Malaysia, Western Indonesia, Thailand and Vietnam. Refer to Table 1 for sampling details. Maps drawn and adapted by Adobe Photoshop CS3 from public domain image provided by D-maps.com (http://d-maps.com/m/asia/asiesudest/asiesudest06.svg).
Morphological identification among adults was not straightforward. Of the adults sampled, a combined total of 14 specimens from Sikao (Isthmus of Kra, Thailand), Alur Itam and Sungai Iyu (Western Indonesia), and Sungai Lalang and Kuala Sepetang (Peninsular Malaysia) could not be classified based on existing keys. At three of these sites (Alur Itam, Sungai Iyu and Kuala Sepetang), additional individuals were present that could be positively identified as D. collettei, so in this case co-occurring unidentified fishes were tentatively classified as being of the same species. In the other two instances (Sikao, n = 6, and Sungai Lalang, n = 12), no positive identifications could be made of any specimen, and therefore all were recorded as Dermogenys sp. No adult male was available from these locations, and although females were morphologically similar to D. collettei, they lacked the thin line of melanophores arranged from the anterior to anal fin characteristic of females of this species.
The remaining 86 (29.7%) samples were sub-adult specimens lacking morphological diagnostic characters, and as sub-adults are expected to co-exist with the adult specimens, where adults could be identified the identity of sub-adults was tentatively assumed to be the same. For all specimens, DNA barcodes were then employed to confirm (or challenge) morphological identifications.
DNA Barcoding for species identification
All samples (290) were successfully DNA barcoded for a 651 bp segment of the mitochondrial COI gene. All sequences have been deposited in GenBank with accession number MG563383 – MG563672. There were no insertions/deletions or stop codons in the alignment, which had mean nucleotide frequencies of A = 23%, T = 31%, C = 27% and G = 19%, and 157 variable nucleotide positions, including 134 parsimoniously informative sites, with most variation occurring at third codon positions. A total of 53 different COI haplotypes were observed among all Dermogenys sequenced.
All sequences were identified as Dermogenys spp. by the GenBank BLAST tool, however, as prior to this study no species-level reference COI data existed for Dermogenys other than for D. pusilla, and as no D. pusilla was collected in this study, no individual could be identified to species level with the GenBank search. The current data set did, however, clearly exhibit variation consistent with five discrete monophyletic groups that all differed from D. pusilla, as visualised in Fig. 2. These groups were largely consistent with species designations based on morphology, identifying groupings for D. collettei (12 haploypes), D. sumatrana (12 haploypes), D. siamensis (15 haploypes), D. bispina (6 haploypes), and a newly observed taxon, Dermogenys sp. (8 haploypes).

COI gene tree of six putative Dermogenys species. Values at nodes represents the bootstrap support and posterior probability (NJ/ML/BI). Gene tree includes sequences retrieved from GenBank for D. pusilla and for outgroups Nomorhamphus sp. (JQ430374) and Hemirhamphodon kuekenthali (KM405787) that were used to root the tree.
Not all was as expected, however, with relatively high genetic distance observed within groups, tentatively identified samples showing identity to unexpected genetic clusters, and individuals collected at the same location showing identity to different groupings. This is reflected in within-site measures of Kimura 2-parameter (K2P) genetic distance, which ranged between 0.0% where all individuals had identical barcodes, to 5.1% where individuals collected together assigned to multiple genetic clusters, indicating more than one putative species was present in these sites (Table 1). Samples from three locations showed high “intrapopulation” K2P distance due to the presence of multiple putative species; Kuala Sepetang (5.1%), Sg. Iyu (3.6%) and Alur Itam (3.8%). Individuals from all three sites had previously been flagged as hard to identify based on morphology, and as a result of the barcode evidence, all specimens from each site were sorted into two groups; the D. collettei group and the Dermogenys sp. group (Table 1) and are considered discrete putative species in all further analysis and discussion.
Average K2P genetic distances among barcodes generated here, Genbank D. pusilla barcodes, and representatives of other zenarchopterids are presented in Table 2. Within putative taxa, as defined using a combination of morphological similarity and genetic monophyly, the lowest mean distance (0.02%) was observed for the unclassified Dermogenys sp. (n = 28), that was collected across five locations (Table 1, Fig. 1). Maximum average distance at the intraspecific level was 1.5% (for D. bispina and D. pusilla), while minimum average between species was 3.7% between D. sumatrana and D. pusilla.
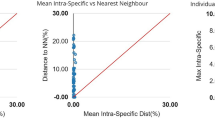
A barcode gap analysis (Fig. 3(a)) incorporating current data and existing D. pusilla barcodes revealed that “barcode gaps” were present among all pairwise comparisons of the six putative Dermogenys species, indicating that all six groups were comprised of members belonging to different putative species. Following this, Automatic Barcode Gap Discovery (ABGD) analysis generated 3 to 48 OTUs (Fig. 4). However, an a priori intraspecific divergence of (P) (P = 0.0077–0.0129), chosen based on Fig. 4, generated 7 OTUs, instead of the expected six. The additional OTU (with respect to the six presumed species) identified by ABGD divided D. bispina into two groups, the first is from Kudat and the second from Sandakan (north and north-east Sabah, respectively).

Maximum intraspecific distance (% K2P) plotted against nearest neighbour distance (% K2P) for the seven OTUs examined in this study. Points above the line indicate species with a barcode gap. (a) Six initial putative morphological species; (b) Newly assigned Dermogenys species groups based on ABGD analysis.

The number of genetically distinct OTUs according to the prior intraspecific divergence value generated by ABGD based on K2P distance. Data are from 299 molecular sequences.
The K2P genetic distances of the newly assigned groupings (based on OTUs) are summarised in Table 3. The minimum genetic distance values of the newly assigned grouping were slightly lower than for the six presumed Dermogenys species, with intraspecific distance ranging between 0.1% - 1.5% and interspecific distance from 2.7% to 13.2%. Average K2P genetic distance generated for the two groups of D. bispina (Kudat and Sandakan) is 2.7%. Overall, the p-distance within-site, intraspecific and intraspecific values were slightly lower than K2P value (Tables 2 and 3). A re-analysis of the barcode gap was conducted for the newly assigned groups, showing the presence of barcode gaps as presented in Fig. 3(b).
The COI gene tree estimated using ML and BI was congruent with that estimated using the NJ method (Fig. 2), with all tree estimation methods yielding results that recover two strongly supported geographic clusters of D. bispina, Kudat and Sandakan, reflecting groups identified in the ABGD analysis. At three locations (Kuala Sepetang (SPP), Sungai Iyu (SI) and Alur Itam(AI)), individuals were present from two taxonomic groups, namely D. collettei and Dermogenys sp.
Discussion
The present study successfully identified and characterized six putative species of Dermogenys, including two reciprocally monophyletic groups in D. bispina; generated a reference barcode database for Dermogenys; and assessed levels of morphological/molecular disparity. Our preliminary Dermogenys taxonomic identification using morphological characters was limited, as the morphological keys11 are only applicable at particular life stages and are gender-specific. Therefore, due to a strong reliance on assessment of anal fin structure, which is only accurate through advanced radiographic analyses, conflict was common between morphologically identified specimens and results from our molecular analysis. These issues compound the mislabelling of Dermogenys species throughout museum collections and the taxonomic literature11,20,57,58,59. Failure to adequately discriminate to species level prior to biological and evolutionary investigations can also be problematic, for example de Bruyn et al.12 found major taxonomic ambiguities in their molecular analysis of Dermogenys, as morphologically recognised species were shown to comprise multiple, reciprocally monophyletic lineages. These taxonomic uncertainties are understandable, given the lack of easily distinguishable diagnostic characters for the group, yet ongoing mislabelling of specimens adds to taxonomic confusion and can lead to counter-productive conservation and management efforts. Thus, methods such as DNA barcoding may offer a means to ensure identifications are standardised, at least until such time as current taxonomic keys receive necessary revisions, alleviating the inconsistencies highlighted above, and the strong reliance on sex-specific characters.
The DNA barcoding approach relies, as to some extent do taxonomic keys, on expert knowledge to ascertain the identify of reference specimens, pulling together taxonomic literature and geographical information to compile a reference library. This paper represents the first attempt to do so for Dermogenys, verifying that the standard fish barcoding primers of Ward et al.31 work for barcoding the genus and adding data for five more Dermogenys species to the existing reference library of one. The DNA barcodes generated here enable the identification not only of adult specimens without undertaking complicated morphological investigations, but also of under-developed sub-adults, in the current example and more importantly, for future researchers seeking to identify Dermogenys.
The use of barcoding has already revealed some new information on geographical distributions of the genus. In clarifying the identity of fishes from Kuala Sepetang, Sg. Iyu and Alur Itam that were all tentatively identified as D. collettei using morphological and geographical information, our survey revealed that two genetically divergent species exist in sympatry at these sites, one of which remains undescribed. Such findings show that DNA barcoding could assess species diversity through the pairing of genetic distance methods and the genotypic cluster concept60. This study also lends further support that DNA barcoding may aid in larval identification, as has been observed in previous studies36,37,38, as well as in the identification of cryptic sympatric species, one of the known strengths of the barcoding approach32,61.
Of all taxa surveyed here, the Dermogenys species from northern Borneo exhibited among the highest intraspecific distance values, with some analyses indicating cause for considering the two geographically and genetically distinct populations, D. bispina ‘Kudat’ (north Borneo) and D. bispina ‘Sandakan’ (northeast Borneo), as sub-species. Borneo boasts a large number of endemic freshwater fishes62,63, and the island’s physical and environmental characteristics, as well as a paleo history of montane regions are believed to be core reasons for the north, in particular, being a centre for speciation and endemism64,65. Between Sandakan and Kudat in northern Borneo lies the Crocker Range (average height 1,800 m) separating eastern and western drainages64,66, and the potential cause for isolating the two D. bispina lineages, leading to formation of subspecies in allopatry. This range has previously been hypothesised as a barrier to gene flow for freshwater fishes in the region67,68.
Minimum distance between the two D. bispina groups was 2.7%, and this would be high enough to qualify as diagnostic of different species if applying a 2% divergence threshold (e.g., Ward et al.30,69. Hubert et al.70). However, the sole reliance of a 2% cut-off value for delimiting species across all taxa can mask the real diversity (e.g., Australian fishes31; Neotropical freshwater fishes34; Canadian freshwater fishes70; Tuna species71; North America’s freshwater fishes72) in the group as the initial intraspecific distance of the D. bispina group (when both Kudat and Sandakan were combined) was only 1.5%, and the initial barcode gap displayed no taxonomic ambiguity. This highlights the need for conducting comprehensive analyses as illustrated in the ABGD analysis. Combined with the lack of clear morphological differences, two OTUs supported with barcode gap re-analysis, the discovery of the two reciprocally monophyletic and geographically isolated D. bispina groups, probably warrants the two populations be considered at the very least as discrete evolutionary significant units or subspecies, and potentially as two different species, given that other reciprocally monophyletic groups with similar levels of divergence are classified as different species (e.g., D. pusilla and D. sumatrana - 3.7%). No overlap of intraspecific and interspecific genetic distance was observed. In fact, lower levels of nearest neighbour distance (NND) were observed in Canadian freshwater fishes70. Out of 190 barcoded species, 14 showed <0.1% NND value, 20 showed 0.1–1.0%, 17 showed 1.0–2.7%. A similar pattern was observed in Nigerian freshwater fishes73. O’Brien & Mayr74 outlined several criteria for subspecies classification 1) subspecies members share unique geographic range or habitat, 2) the OTUs are reciprocally monophyletic indicating that the genetic divergence of subspecies accumulated in the absence of gene flow, and is time-dependent and 3) unique natural history relative to other subdivisions of the species.
Even though DNA barcoding is a very effective tool for the systematics and validation of numerous freshwater fish taxa32,34,70,75, this approach leans heavily on the work of classical taxonomists, including the primary documentation of species and distributions, and ongoing work in validating and describing new OTUs. Morphological identification is likely to remain a fundamental approach for taxonomic identifications in most instances, and where DNA barcodes find no match in the barcode libraries, morphology remains the first port of call to validate a specimen’s identity76. Nevertheless, ambiguity in molecular findings is a good indication that knowledge of a taxonomic group is incomplete, and thus, DNA barcoding functions as a complementary and supporting tool for the robust identification of fish taxa and other organisms.
Conclusion
This study reinforces the complementarity of both morphological and molecular characters as well as other lines of evidence (geographical distribution) in elucidating the taxonomic status and systematics of the Dermogenys group. The data presented contributes DNA reference barcodes for five additional species in this taxonomically challenging group, and we shed light on the level of genetic divergence expected within and between species in this genus, highlighting an area of Northern Borneo where geographically distinct lineages have arisen below the recognised species boundary. Taxonomy and systematics of the group remains incomplete, and detailed taxonomic work will be required to formally describe the new OTU (designated here as Dermogenys sp.), and to update taxonomic keys accordingly. Never-the-less, DNA barcoding as employed here demonstrates the power of molecular techniques in helping tackle difficult issues in taxonomy.
Methods
Collection of tissue samples
This study was carried out in accordance with the recommendations and approval by the Universiti Sains Malaysia Animal Ethics Committee. A total of 288 individuals from 31 locations were sampled from the inland waters of Malaysia, Western Indonesia, Thailand and Vietnam (Fig. 1, Table 1). Samples were obtained from slow moving brackish and freshwater systems, and collected using a scoop net with mesh size of 4 mm. The specimens were identified to genus level based on Rainboth20. Specimens were anesthetized with Transmore (NIKA Trading Co.), a fish stabilizer commonly used in aquatic trading prior to taking tissue samples from the pectoral fin (stored in 95% ethanol for DNA extraction). The specimens were then fixed with formalin and preserved in 70% ethanol. Back at the laboratory species level identification was conducted based on osteological characters of the modified anal fin (andropodium) in males11. The radiological study was conducted at the Chinese Academy of Sciences, Beijing. Preliminary identification was conducted based on morphological keys. As many characters are common in groups of species (e.g. melanophores from anterior to anal fin arranged into thin line observed in D. collettei and D. siamensis females; black pigment on distal tips of the posterior dorsal fin present in D. collettei, D. siamensis, Dermogenys sp. and D. bispina males; black pigment at the base and distal tip of pelvic fin observed in D. collettei, D. sumatrana, D. siamensis, and Dermogenys sp. males), precise identifications considered current knowledge on species distributions1,11,16,77. Thus, samples from Sabah, Vietnam, Thailand and Western Indonesia were putatively assigned to D. bispina, D. siamensis and D. sumatrana respectively, as a working hypothesis. Although de Bruyn et al.12 did not identify their specimens to species level, they hypothesised that most species were restricted in distribution range, which was further supported by their phylogeny, and therefore this classification scheme was justified in our study. In addition, two samples - morphologically identified as D. collettei, were contributed by the Florida Museum of Natural History (DC652 and DC683), making a total of 290 samples from 33 sampling sites (Fig. 1).
Extraction, COI amplification and DNA sequencing
Total genomic DNA was extracted by using a modified conventional salt extraction procedure78. The template DNA was amplified by PCR in a 25 μL mixture containing 2.0 μL of DNA, 2.5 μL 10 × PCR buffer, 3.3 μL of 25 mM MgCl2, 0.5 μL 10 mM dNTPs, 0.25 μL 10 μM primers, 0.1 μL i-Taq plus polymerase and 16.1 μL DNAse-free water. The primers used for the amplification of the COI gene31 were Fish-F2 5′-TCG ACT AAT CAT AAA GAT ATC GGC AC-3′ and Fish-R2 5′-ACT TCA GGG TGA CCG AAG AAT CAG AA-3′. Amplifications were performed using a BioRad Thermocycler at 94 °C initial denaturation and 34 cycles with the following conditions: 20 s at 94 °C, 20 s at 47.9 °C and 70 s at 72 °C. The PCR products were sent for sequencing to 1st BASE Sequencing Service Sdn. Bhd. (Malaysia).
Sequence analysis
Sequences were aligned using MEGA v6.0679 software, this package was also used to investigate base composition, and to calculate the number of variable sites and genetic distance measures. Both uncorrected pairwise distance (p-distance) and distance estimates based on the Kimura 2-parameter (K2P) model80 were calculated to assess mean “intrapopulation”, intraspecific and interspecific genetic distance within sample sites, and within and between species. Using the same software, phylogenetic relationships among haplotypes were constructed applying Neighbour-Joining (NJ) and Maximum Likelihood (ML) methods with 1000 bootstrap replicates. Tree construction was conducted using the K2P model80 for NJ analysis and Hasegawa-Kishino-Yano model81 with gamma (HKY + G) rates (optimal substitution model estimated by the model test run in MEGA v6.06) for ML analysis.
The relationships among haplotypes were also assessed using a Bayesian Inference (BI) method together with a Markov Chain Monte Carlo (MCMC) algorithm. In order to construct gene trees, PartitionFinder v1.1.082 was used to determine the best-fit partitioning schemes and models of molecular evolution for phylogenetic analysis. The BI analyses were performed using MrBayes83 with employment of HKY + I81, F8184, and GTR + G85 for the first, second and third codon, 1 million MCMC chains and a 50% burn in. The trace files generated from MrBayes run were diagnosed in Tracer v1.686 to evaluate the MCMC chain. The COI trees generated were visualized and edited using FigTree v1.4.287. Nomorhamphus sp. (GenBank Acc. No JQ430374) and Hemirhamphodon (Acc. No KM405787) were included as outgroups88. Additional D. pusilla (Acc. KU692464 to KU692472) COI sequences from GenBank were included to improve phylogenetic resolution.
The maximum intraspecific distance against the minimum nearest-neighbour distance graph was plotted to check the presence of a “barcode gap” in the dataset89. Presence of barcode gaps among sets of sequences within a presumed species/taxon indicates that there is likely to be more than a single taxon within the group. The number of operational taxonomic units (OTUs) based on pairwise sequence distances between individuals within the dataset was generated using the Automatic Barcode Gap Discovery (ABGD)90 species delineation tool on a web interface (http://wwwabi.snv.jussieu.fr/public/abgd/abgdweb.html) with default settings and the K2P model employed. The interpretation of the ABGD results is very straightforward. An OTU is considered as successfully delimited when the predicted groups are formed and no other unrelated sequences were included in that group91.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Meisner, A. D. & Collette, B. B. A new species of viviparous halfbeak, Dermogenys bispina (Teleostei: Hemiramphidae) from Sabah (North Borneo). Raffles Bulletin of Zoology 46, 373–380 (1998).
Stolk, A. Histo-endocrinological analysis of gestation phenomena in the Hemiramphid Dermogenys pusillus Van Hasselt. IV. Corpus luteum cycle during pregnancy. Acta Morphologica Neerlando-Scandinavica 1, 209–223 (1956).
Stolk, A. Role of cell division in gastrulation of the hemiramphid Dermogenys pusillus van hasselt. Cellular and Molecular Life Sciences 18, 367–367 (1962).
Forward, R. B. Jr & Waterman, T. H. Evidence for E-vector and light intensity pattern discrimination by the teleost. Dermogenys. Journal of Comparative Physiology 87, 189–202 (1973).
Flegler, C. Electron microscopic studies on the development of the chorion of the viviparous teleost Dermogenys pusillus (Hemirhamphidae). Cell and Tissue Research 179, 255–270 (1977).
Downing, A. L. & Burns, J. R. Testis morphology and spermatozeugma formation in three genera of viviparous halfbeaks: Nomorhamphus, Dermogenys, and Hemirhamphodon (Teleostei: Hemiramphidae). Journal of Morphology 225, 329–343 (1995).
Clemen, G., Wanninger, A. C. & Greven, H. The development of the dentigerous bones and teeth in the hemiramphid fish Dermogenys pusillus (Atheriniformes, Teleostei). Annals of Anatomy-Anatomischer Anzeiger 179, 165–174 (1997).
Greven, H., Wanninger, A. C. & Clemen, G. Dentigerous bones and dentition in the hemiramphid fish Dermogenys pusillus (Atheriniformes, Teleostei). Annals of Anatomy-Anatomischer Anzeiger 179, 21–32 (1997).
Meisner, A. D. & Burns, J. R. Viviparity in the halfbeak genera Dermogenys and Nomorhamphus (Teleostei: Hemiramphidae). Journal of Morphology 234, 295–317 (1997).
Meisner, A. D. & Burns, J. R. Testis and andropodial development in a viviparous halfbeak, Dermogenys sp. (Teleostei: Hemiramphidae). Copeia, 44–52 (1997).
Meisner, A. D. Phylogenetic systematics of the viviparous halfbeak genera Dermogenys and Nomorhamphus (Teleostei: Hemiramphidae: Zenarchopterinae). Zoological Journal of the Linnean Society 133, 199–283 (2001).
de Bruyn, M. et al. Paleo-drainage basin connectivity predicts evolutionary relationships across three Southeast Asian biodiversity hotspots. Systematic Biology 62, 398–410 (2013).
Zanger, K. & Greven, H. The “nasal barbel” of the halfbeak Dermogenys pusilla (Teleostei: Zenarchopteridae)–an organ with dual function. Vertebrate. Zoology 63, 183–191 (2013).
Ho, J. K. et al. Shoaling behaviour in the pygmy halfbeak Dermogenys collettei (Beloniformes: Zenarchopteridae): comparing populations from contrasting predation regimes. Raffles Bulletin of Zoology 63, 237–245 (2015).
Myers, N., Mittermeier, R. A., Mittermeier, C. G., Da Fonseca, G. A. & Kent, J. Biodiversity hotspots for conservation priorities. Nature 403(6772), 853–858 (2000).
Brooks, T. M. et al. Habitat loss and extinction in the hotspots of biodiversity. Conservation biology 16(4), 909–923 (2002).
van Hasselt, J. C. Uittreksel uit een’brief van Dr. JC van Hasselt aan Professor van Swinderen. Algemeene Konst-en Letter-Bode, 230-233 (1823).
Mohr, E. R. N. A. Hemirhamphiden Studien. IV. Die Gattung Dermogenys van Hasselt. V. Die Gattung Nomorhamphus Weber & de Beaufort. VI. Die Gattung Hemirhamphodon Bleeker. Mitt. zool. Mus. Berlin 21, 34–64 (1936).
Brembach, M. Lebendgebärende Halbschnäbler. (Verlag Natur und Wissenschaft, Solingen, 1991).
Rainboth, W. J. Fishes of the Cambodian Mekong. (Food & Agriculture Org., 1996).
Melvin, G. D., Dadswell, M. J. & McKenzie, J. A. Usefulness of meristic and morphometric characters in discriminating populations of American shad (Alosa sapidissima)(Ostreichthyes: Clupeidae) inhabiting a marine environment. Canadian Journal of Fisheries and Aquatic Sciences 49, 266–280 (1992).
Haddon, M. & Willis, T. J. Morphometric and meristic comparison of orange roughy (Hoplostethus atlanticus: Trachichthyidae) from the Puysegur Bank and Lord Howe Rise, New Zealand, and its implications for stock structure. Marine Biology 123, 19–27 (1995).
Rainboth, W. J. The taxonomy, systematics, and zoogeography of Hypsibarbus, a new genus of large barbs (Pisces, Cyprinidae) from the rivers of Southeastern Asia. Vol. 129 (Univ of California Press, 1996).
Murta, A. G. Morphological variation of horse mackerel (Trachurus trachurus) in the Iberian and North African Atlantic: implications for stock identification. ICES Journal of Marine Science: Journal du Conseil 57, 1240–1248 (2000).
Turan, C. Stock identification of Mediterranean horse mackerel (Trachurus mediterraneus) using morphometric and meristic characters. ICES Journal of Marine Science: Journal du Conseil 61, 774–781 (2004).
Turan, C., Oral, M., Öztürk, B. & Düzgüneş, E. Morphometric and meristic variation between stocks of Bluefish (Pomatomus saltatrix) in the Black, Marmara, Aegean and northeastern Mediterranean Seas. Fisheries Research 79, 139–147 (2006).
Hossain, M. A., Nahiduzzaman, M., Saha, D., Khanam, M. U. H. & Alam, M. S. Landmark-based morphometric and meristic variations of the endangered Carp, Kalibaus Labeo calbasu, from stocks of two isolated rivers, the Jamuna and Halda, and a hatchery. Zoological Studies 49, 556–563 (2010).
Khan, M. A., Miyan, K. & Khan, S. Morphometric variation of snakehead fish, Channa punctatus, populations from three Indian rivers. Journal of Applied Ichthyology 29, 637–642 (2013).
Chakona, A., Swartz, E. R. & Skelton, P. H. A new species of redfin (Teleostei, Cyprinidae, Pseudobarbus) from the Verlorenvlei River system, South Africa. ZooKeys, 121–137 (2014).
González-Castro, M. & Ghasemzadeh, J. In Biology, Ecology and Culture of Grey Mullet (Mugilidae), Biology, Ecology and Culture of Grey Mullet (Mugilidae) (ed Crosetti D. & Blaber S. J. M.) 1–22 (CRC Press, 2015).
Ward, R. D., Zemlak, T. S., Innes, B. H., Last, P. R. & Hebert, P. D. DNA barcoding Australia’s fish species. Philosophical Transactions of the Royal Society of London B: Biological Sciences 360, 1847–1857 (2005).
Lara, A. et al. DNA barcoding of Cuban freshwater fishes: evidence for cryptic species and taxonomic conflicts. Molecular Ecology Resources 10, 421–430 (2010).
Lakra, W. S. et al. DNA barcoding Indian marine fishes. Molecular Ecology Resources 11, 60–71 (2011).
Pereira, L. H., Hanner, R., Foresti, F. & Oliveira, C. Can DNA barcoding accurately discriminate megadiverse Neotropical freshwater fish fauna? BMC Genetics 14, 1 (2013).
Geiger, M. F. et al. Spatial heterogeneity in the Mediterranean Biodiversity Hotspot affects barcoding accuracy of its freshwater fishes. Molecular Ecology Resources 14, 1210–1221 (2014).
Webb, K. E., Barnes, D. K., Clark, M. S. & Bowden, D. A. DNA barcoding: a molecular tool to identify Antarctic marine larvae. Deep Sea Research Part II: Topical Studies in Oceanography 53, 1053–1060 (2006).
Victor, B. C., Hanner, R., Shivji, M., Hyde, J. & Caldow, C. Identification of the larval and juvenile stages of the Cubera Snapper, Lutjanus cyanopterus, using DNA barcoding. Zootaxa 2215, 24–36 (2009).
Hubert, N., Delrieu-Trottin, E., Irisson, J. O., Meyer, C. & Planes, S. Identifying coral reef fish larvae through DNA barcoding: a test case with the families Acanthuridae and Holocentridae. Molecular Phylogenetics and Evolution 55, 1195–1203 (2010).
Wong, E. H. K. & Hanner, R. H. DNA barcoding detects market substitution in North American seafood. Food Research International 41, 828–837 (2008).
Barbuto, M. et al. DNA barcoding reveals fraudulent substitutions in shark seafood products: the Italian case of “palombo”(Mustelus spp. Food Research International 43, 376–381 (2010).
Galimberti, A. et al. DNA barcoding as a new tool for food traceability. Food Research International 50, 55–63 (2013).
Chin, T. C., Adibah, A. B., Hariz, Z. D. & Azizah, M. S. Detection of mislabelled seafood products in Malaysia by DNA barcoding: Improving transparency in food market. Food Control 64, 247–256 (2016).
Ball, S. L. & Armstrong, K. F. DNA barcodes for insect pest identification: a test case with tussock moths (Lepidoptera: Lymantriidae). Canadian Journal of Forest Research 36, 337–350 (2006).
Nagoshi, R. N., Brambila, J. & Meagher, R. L. Use of DNA barcodes to identify invasive armyworm Spodoptera species in Florida. Journal of Insect Science 11, 1–11 (2011).
Marko, P. B. et al. Fisheries: mislabelling of a depleted reef fish. Nature 430, 309–310 (2004).
Ogden, R., Dawnay, N. & McEwing, R. Wildlife DNA forensics - bridging the gap between conservation genetics and law enforcement. Endangered Species Research 9, 179–195 (2009).
Dalton, D. L. & Kotze, A. DNA barcoding as a tool for species identification in three forensic wildlife cases in South Africa. Forensic Science International 207, e51–e54 (2011).
Sati, J. et al. Phylogenetic relationship and molecular identification of five Indian Mahseer species using COI sequence. Journal of Environmental Biology 34, 933 (2013).
Schoelinck, C., Hinsinger, D. D., Dettaï, A., Cruaud, C. & Justine, J. L. A phylogenetic re-analysis of groupers with applications for ciguatera fish poisoning. PloS One 9, e98198 (2014).
Prehadi, S. A., Kurniasih, E. M., Rahmad, A. D., Subhan, B. & Madduppa, H. H. DNA barcoding and phylogenetic reconstruction of shark species landed in Muncar fisheries landing site in comparison with Southern Java fishing port. Biodiversitas 16, 55–61 (2015).
Rubinoff, D. Utility of mitochondrial DNA barcodes in species conservation. Conservation Biology 20, 1026–1033 (2006).
Ardura, A., Linde, A. R., Moreira, J. C. & Garcia-Vazquez, E. DNA barcoding for conservation and management of Amazonian commercial fish. Biological Conservation 143, 1438–1443 (2010).
Hardy, C. M., Adams, M., Jerry, D. R., Morgan, M. J. & Hartley, D. M. DNA barcoding to support conservation: species identification, genetic structure and biogeography of fishes in the Murray—Darling River Basin, Australia. Marine and Freshwater Research 62, 887–901 (2011).
Iyengar, A. Forensic DNA analysis for animal protection and biodiversity conservation: a review. Journal for Nature Conservation 22, 195–205 (2014).
Bleeker, P. Zevende bijdrage tot de kennis der ichthyologische fauna van Borneo. Zoetwatervisschen van Sambas, Pontianak en Pangaron. Natuurkundig Tijdschrift voor Nederlandsch Indie¨ 5, 427–462 (1853).
Fowler, H. W. Zoological results of the third De Schauensee Siamese Expedition, Part I: Fishes. Proceedings of the Academy of Natural Sciences of Philadelphia 86, 67–163 (1934).
Doi, A. et al. A collection of freshwater fishes from the Rayu basin of western Sarawak, Malaysia. Raffles Bulletin of Zoology 49, 13–18 (2001).
Roos, N., Chamnan, C., Loeung, D., Jakobsen, J. & Thilsted, S. H. Freshwater fish as a dietary source of vitamin A in Cambodia. Food Chemistry 103, 1104–1111 (2007).
Hung, L. T., Luong, V. C.., Hoa, N. P. & Diana, J. in Better science, better fish, better life: Proceedings of the ninth international symposium on tilapia in aquaculture (eds Liping, L. & Fitzsimmons, K.), section II, 88–100 (AquaFish Collaborative Research Support Program, 2011).
Mallet, J. A species definition for the modern synthesis. Trends in Ecology & Evolution 10, 294–299 (1995).
Valdez-Moreno, M., Vásquez-Yeomans, L., Elías-Gutiérrez, M., Ivanova, N. V. & Hebert, P. D. Using DNA barcodes to connect adults and early life stages of marine fishes from the Yucatan Peninsula, Mexico: potential in fisheries management. Marine and Freshwater Research 61, 655-671 (2010).
Kottelat, M. & Whitten, T. Freshwater biodiversity in Asia with special reference to fish. Vol. 343 (World Bank Publications, 1996).
Yap, S. Y. On the distributional patterns of Southeast‐East Asian freshwater fish and their history. Journal of Biogeography 29, 1187–1199 (2002).
Sheldon, F. H., Moyle, R. G. & Kennard, J. Ornithology of Sabah: history, gazetteer, annotated checklist and bibliography. No. 52 (American Ornithologists’ Union, 2001).
Das, I. Crocker Range National Park, Sabah, as a refuge for Borneo’s montane herpetofauna. Amphibian and Reptile Conservation 4, 3–11 (2006).
Simons, W. J. F. et al. A decade of GPS in Southeast Asia: Resolving Sundaland motion and boundaries. Journal of Geophysical Research: Solid Earth 112, B06420 (2007).
Yano, K., et al. Sharks and rays of Malaysia and Brunei Darussalam. (Marine Fishery Resources Development and Management Department, Southeast Asian Fisheries Development Center, 2005).
Chong, V. C., Lee, P. K. Y. & Lau, C. M. Diversity, extinction risk and conservation of Malaysian fishes. Journal of Fish Biology 76, 2009–2066 (2010).
Ward, R. D., Hanner, R. & Hebert, P. D. The campaign to DNA barcode all fishes, FISH‐BOL. Journal of Fish Biology 74, 329–356 (2009).
Hubert, N. et al. Identifying Canadian freshwater fishes through DNA barcodes. PLoS One 3(6), e2490 (2008).
Lowenstein, J. H., Amato, G. & Kolokotronis, S. O. The real maccoyii: identifying tuna sushi with DNA barcodes–contrasting characteristic attributes and genetic distances. PLoS One 4, e7866 (2009).
April, J., Mayden, R. L., Hanner, R. H. & Bernatchez, L. Genetic calibration of species diversity among North America’s freshwater fishes. Proceedings of the National Academy of Sciences 108, 10602–10607 (2011).
Nwani, C. D. et al. DNA barcoding discriminates freshwater fishes from southeastern Nigeria and provides river system-level phylogeographic resolution within some species. Mitochondrial DNA 22(sup1), 43–51 (2011).
O’Brien, S. J. & Mayr, E. Bureaucratic mischief: recognizing endangered species and subspecies. Science(Washington) 251, 1187–1188 (1991).
Lim, H., Abidin, M. Z., Pulungan, C. P., de Bruyn, M. & Nor, S. A. M. DNA barcoding reveals high cryptic diversity of the freshwater halfbeak genus Hemirhamphodon from Sundaland. PloS one 11, e0163596 (2016).
Hajibabaei, M., Singer, G. A., Clare, E. L. & Hebert, P. D. Design and applicability of DNA arrays and DNA barcodes in biodiversity monitoring. BMC Biology 5, 1–7 (2007).
Kottelat, M. The fishes of the inland waters of Southeast Asia: a catalogue and core bibliography of the fishes known to occur in freshwaters, mangroves and estuaries. Raffles Bulletin of Zoology, 1–663 (2013).
Watts, P. Extraction of DNA from tissue: High salt method. University of Liverpool https://www.liverpool.ac.uk/~kempsj/IsolationofDNA.pdf (2001).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular Biology and Evolution 30, 2725–2729 (2013).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution 16, 111–120 (1980).
Hasegawa, M., Kishino, H. & Yano, T. Dating of human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22, 160–174 (1985).
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T. & Calcott, B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Molecular Biology and Evolution 34, 772–773 (2016).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Felsenstein, J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376 (1981).
Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences in Some Mathematical Questions In Biology: DNA Sequence Analysis (ed. Miura, R. M.) 57-86 (American Mathematical Society, 1986).
Rambaut, A., Suchard, M. A., Xie, D. & Drummond, A. J. Tracer (version1.6) at http://beast.bio.ed.ac.uk (2015).
Rambaut, A. FigTree: Tree figure drawing tool (version 1.4.2) at http://ac.uk/software/figtree/ (2015).
Lovejoy, N. R., Iranpour, M. & Collette, B. B. Phylogeny and jaw ontogeny of beloniform fishes. Integrative and Comparative Biology 44, 366–377 (2004).
Meyer, C. P. & Paulay, G. DNA barcoding: error rates based on comprehensive sampling. PLoS biology 3, 2229–2238 (2005).
Puillandre, N., Lambert, A., Brouillet, S. & Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Molecular Ecology 21, 1864–1877 (2012).
Krishnamurthy, P. K. & Francis, R. A. A critical review on the utility of DNA barcoding in biodiversity conservation. Biodiversity and Conservation 21(8), 1901–1919 (2012).
Acknowledgements
Samples from Western Indonesia, Thailand and Vietnam were obtained in collaboration with local university researchers. Radiography was conducted at the Chinese Academy of Sciences, Beijing. This study was funded by Universiti Sains Malaysia, IReC grant (1002/PBIOLOGI/910403) and FRGS grant (203/PBIOLOGI/6711522).
Author information
Authors and Affiliations
Contributions
S.N.F. collected material in the field, conducted the experiments, data analysis, and authored the manuscript; Z.A.M., T.Y.D. and T.S. made this project possible by facilitating sampling and contributing ideas; L.M.P. provided several materials and contributed to morphological identification; Y.Z. contributed to radiological analysis; E.A.S.A. provided reassurance on taxonomic identifications, advised on data analysis and co-wrote the manuscript; M.Z.K. contributed to the morphological identification; M.d.B. and M.N.S.A. supervised S.N.F. and contributed to writing the manuscript; M.N.S.A. provided laboratory support.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nurul Farhana, S., Muchlisin, Z.A., Duong, T.Y. et al. Exploring hidden diversity in Southeast Asia’s Dermogenys spp. (Beloniformes: Zenarchopteridae) through DNA barcoding. Sci Rep 8, 10787 (2018). https://doi.org/10.1038/s41598-018-29049-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29049-7
This article is cited by
-
DNA Barcoding for Identification and Discovery of Fish Species in the Protected Mangroves of Hormozgan, Iran
Estuaries and Coasts (2023)
-
Reassessing fish diversity of Penang Island’s freshwaters (northwest Peninsular Malaysia) through a molecular approach raises questions on its conservation status
Biodiversity and Conservation (2022)
-
Disentangling the taxonomy of the subfamily Rasborinae (Cypriniformes, Danionidae) in Sundaland using DNA barcodes
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}