
Abstract
Chloroplast genomes of plants are highly conserved in both gene order and gene content, are maternally inherited, and have a lower rate of evolution. Chloroplast genomes are considered to be good models for testing lineage-specific molecular evolution. In this study, we use Schisandraceae as an example to generate insights into the overall evolutionary dynamics in chloroplast genomes and to establish the phylogenetic relationship of Schisandraceae based on chloroplast genome data using phylogenomic analysis. By comparing three Schisandraceae chloroplast genomes, we demonstrate that the gene order, gene content, and length of chloroplast genomes in Schisandraceae are highly conserved but experience dynamic evolution among species. The number of repeat variations were detected, and the Schisandraceae chloroplast genome was revealed as unusual in having a 10 kb contraction of the IR due to the genome size variations compared with other angiosperms. Phylogenomic analysis based on 82 protein-coding genes from 66 plant taxa clearly elucidated that Schisandraceae is a sister to a clade that includes magnoliids, monocots, and eudicots within angiosperms. As to genus relationships within Schisandraceae, Kadsura and Schisandra formed a monophyletic clade which was sister to Illicium.
Similar content being viewed by others

Introduction
Chloroplasts are the photosynthetic organelle that provides energy for plants. The chloroplast has its own genome. In angiosperms, most chloroplast genomes are composed of circular DNA molecules ranging from 120 to 160 kb in length and have a quadripartite organization consisting of two copies of inverted repeats (IRs) of approximately 20–28 kb in size, which divide the rest of chloroplast genome into an 80–90 kb large single copy (LSC) region and a 16–27 kb small single copy (SSC) region. Additionally, the chloroplast genome encodes approximately 114 genes, including four ribosomal RNA (rRNAs), 30 transfer RNA (tRNAs), and approximately 80 unique proteins. Chloroplast protein-coding genes are involved in major functions, which include components of the photosynthetic machinery (such as photosystem I (PSI), photosystem II (PSII), the cytochrome b6/f complex, and the ATP synthase), transcription, and translation.
In general, the chloroplast genome has conserved genome structure, gene content and gene order in most angiosperm plants1,2. However, structural rearrangements, gene loss, IR expansion and inversion occur in certain lineages. In parasitic plants, pseudogenization, gene deletions, and intron losses commonly occur during chloroplast genome evolution3. Some angiosperm plant groups are amenable to large-scale rearrangements, include Campanulaceae4,5,6, Geraniaceae7,8,9 and some legume family species10. A pair of large IR could stabilize the chloroplast genome against major structural rearrangements11,12. Extensions or contractions of IR regions, gene loss and intron loss also commonly occur during chloroplast genome evolution in angiosperms13,14.
In the chloroplast genome, microstructural mutations such as indels, small inversions, and inverted repeats have provided valuable resources to research genome evolution among plants15. Additionally, the chloroplast genome contains more repeated sequences, including simple sequence repeats (SSR), short tandem repeats (STR), homopolymeric repeats, and long repeats, which are assumed to have originated from different mechanisms such as gene conversion, intramolecular recombination, and slipped-strand mispairing (SSM)16. Repeated sequences are also the main resources for genomic events of duplication, deletion, and rearrangement in chloroplast genomes15,17.
Due to maternal inheritance and the rate of evolution, chloroplast genome sequences have long been a focus of research in plant phylogeographic and molecular evolution, as well as phylogenetic, phylogenomic, and genome evolution14. As a result of these characteristics, chloroplast genomes are considered good models for testing lineage-specific molecular evolution. For example, in recent years, the complete chloroplast genome as a super-barcode has gained popularity because it provides more information leading to greatly increased resolution at lower plant taxonomic levels18,19. SNP and indels were other particularly informative for population and biogeography studies20. The development of next-generation sequencing (NGS) and also the third-generation sequencer have provided scientists with faster and less expensive approaches to sequence chloroplast genomes21. Schisandraceae is a small family of the order Austrobaileyales consisting of three genera: Schisandra Michx. with approximately 25 species, Kadsura Kaempf. ex Juss. with approximately 22 species, and Illicium L. with approximately 42 species22. The majority of Schisandraceae species are distributed in temperate and subtropical forests in Southeast Asia and North America22,23. Several species of Schisandraceae have been used in traditional Chinese medicine for many years for the purposes of increasing physical working capacity, relieving pain, and treating skin inflammation24,25.
The classification systems before APG II segregated the genus Illicium as a distinct family, Illiciaceae, and Schisandra and Kadsura, in the family Schisandraceae23,26,27. In addition, the infra-generic classifications in Schisandraceae are still unstable, while molecular phylogenetic analyses concluded that neither Schisandra nor Kadsura is monophyletic24. Therefore, DNA markers with higher resolution are in need for better determining the unresolved lineages in Schisandraceae. Schisandraceae is one of the earliest diverging lineages in angiosperms, however its chloroplast genome evolved in a unique manner to have a 10 kb contraction of the IR. Previous studies had already discovered this phenomenon in genus Illicium and Schisandra28,29. Whether this pattern stays stable in the family needs further examination.
In the present study, we reconstructed the whole chloroplast genome of Kadsura coccinea by using next-generation sequencing and further integrated the available Illicium and Schisandra chloroplast genomes of Schisandraceae. Hence, every genus of Schisandraceae has its representative species present in this study. The objectives of this study were (1) to establish and characterize the organization of the complete chloroplast genome of Kadsura coccinea; (2) to gain in-depth insights into the overall evolutionary dynamics of Schisandraceae chloroplast genomes; and (3) to calibrate the phylogenetic position of Schisandraceae based on phylogenomic analysis.
Materials and Methods
Taxon sampling, DNA extraction and sequencing
Fresh leaves of Kadsura coccinea were collected from a tree at the Research Institute of Forestry, Chinese Academy of Forestry. The fresh leaves were immediately dried with silica gel before DNA extraction. Total DNA was extracted from approximately 10 g of leaves through an improved method by Li et al.30. The quality of DNA was determined by a Nanodrop-2000 spectrometer (Nanodrop Technologies, Wilmington, DE, USA) and agarose gel electrophoresis. DNA was randomly fragmented into 400–600 bp using an ultrasonicator. An Illumina paired-end DNA library was constructed using the NEBNext® Ultra™DNA Library Prep Kit following the manufacturer’s instructions. Paired-end sequencing (2 × 150 bp) was carried out on an Illumina HiSeq. 4000 platform.
Genome assembly and genome annotation
The paired-end reads were qualitatively assessed and initially assembled with SPAdes 3.6.131. Contigs of low sequencing depths were discarded. The remaining contigs may contain the information not only from the chloroplast genome but also from the nuclear genome and the mitochondrial genome. Next, chloroplast genome sequence contigs were selected by performing a BLAST search with default parameters using the Schisandra chinensis chloroplast genome sequence as a reference (GenBank accession number: KU362793)32. Then, the selected contigs were assembled with Sequencher 5.4.5 (Gene Codes, Ann Arbor, MI). The gaps between the plastomic contigs or ambiguous nucleotides were closed by obtaining amplicons with specific primers and directly sequencing the amplicons. The four junctions between the inverted repeats (IRs) and small single copy (SSC)/large single copy (LSC) regions were confirmed with PCR-based product sequencing2.
Chloroplast genome annotation was performed with Plann33 using the Schisandra chinensis reference sequence from GenBank. A chloroplast genome map was drawn using Genome Vx software34. The complete chloroplast genome sequence was deposited in GenBank.
Repeat sequence and SSR element analyses
The size and location of repeat sequences, including forward and palindromic repeats, within the chloroplast genome of Illicium oligandrum, Kadsura coccinea and Schisandra chinensis were identified using REPuter software35. The repeats were identified according to the following conditions: (1) hamming distance of 3, (2) sequence identity ≥90%, and (3) minimum repeat size ≥30 bp. Tandem repeats were identified using web-based Tandem Repeats Finder (https://tandem.bu.edu/trf/trf.html), with 2, 7, and 7 set for the alignment parameters match, mismatch, and indel, respectively. SSRs in the chloroplast genome were detected using MISA (MIcroSAtellite; http://pgrc.ipk-gatersleben.de/misa) with the parameters set at >10 for mononucleotide, >5 for dinucleotide, >4 for trinucleotide, and >3 for tetranucleotide, pentanucleotide, and hexanucleotide SSRs.
Comparative genome analysis
Genome structures among the three genera of the Schisandraceae were compared using mVISTA software in Shuffle-LAGAN mode36. Illicium oligandrum was set as a reference. Subsequently, the nucleotide diversity of the chloroplast genome was conducted based on a sliding window analysis with the DnaSP v5.10 software37. The step size was set to 200 base pairs, and the window length was set to 800 base pairs. The chloroplast genome borders of LSC, SSC, and IRs were compared according to their annotations.
Phylogenetic analyses
To examine the phylogenetic position of Schisandraceae in angiosperms and the relationship among genus in Schisandraceae, 59 complete chloroplast genomes representing the lineages of angiosperms, especially early angiosperms, were downloaded from NCBI Organelle Genome Resource database. GenBank information for all of the chloroplast genomes used for the present phylogenetic analyses can be found in Supplementary Table S2.
The 82 shared protein-coding gene sequences were extracted using a Python script and aligned separately by MAFFT v738. The alignment was manually adjusted, and the specific indels were deleted from the sequences. Phylogenetic trees were reconstructed based on 82 concatenated protein-coding gene sequences by maximum likelihood (ML) and Bayesian inference (BI) methods.
The best-fitting model of sequence evolution was identified with ModelFinder39 based on the Akaike Information Criterion (AIC). Maximum likelihood (ML) analysis was performed using the IQ-TREE V1.6.1 software package40 with 500 non-parametric bootstrap replicates.
Bayesian inference (BI) was performed with MrBayes 3.2.241. Two independent Markov chain Monte Carlo (MCMC) chains, each with three heated and one cold chain, were run for 5 million generations. Each chain started with a random tree, default priors, and sampling trees every 1,000 generations. The MCMC convergence was assumed when the average standard deviation of split frequencies reached 0.01 or less. The first 25% of trees from all runs were discarded as burn-in, and the remaining trees were used to construct majority-rule consensus trees.
Accession code
Kadsura coccinea chloroplast genome are available in GenBank database (accession number: MH029822).
Results
Genome content and organization
A total of approximately 5.2 Gb of 150 bp pair-end reads for Kadsura coccinea were obtained from the Illumina paired-end sequencing, and the reads were then trimmed and assembled using the SPAdes assembler pipeline. The de novo assembled contigs were searched against the chloroplast genome sequences of Schisandra chinensis; eleven contigs were retained. Gaps between contigs were obtained with amplicons from PCR procedures. The total reads were re-mapped to the chloroplast genome, and correction of the sequences was confirmed. Four junction regions of chloroplast genomes were validated using PCR-based sequencing. The coverage of the chloroplast genome was 1233 X, and the sequence of the chloroplast genome was registered into GenBank with the accession number MH029822.
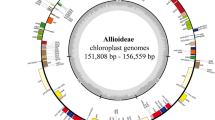
Similar to other higher plants, the chloroplast genome of Kadsura coccinea had a typical quadripartite structure with two inverted repeats (each 16,536 bp in length) separated by one small single-copy region and one large single-copy region (18,040 and 94,301 bp in length, respectively) (Fig. 1). The chloroplast genome of Kadsura coccinea was 145,413 bp in length (Fig. 1). The overall GC content of the chloroplast DNA was 39.7%. The GC content was 38.9%, 35.0%, and 45.5% in the LSC, SSC, and IR regions, respectively. The high GC contents in the IR regions are mainly due to the high GC contents of the four ribosomal RNA (rRNA) genes. Among the representative Schisandraceae species, Kadsura coccinea exhibits the smallest genome size compared with the other two chloroplast genomes. The genome of Illicium oligandrum (148,553 bp) is approximately 3.1 kb larger than that of Kadsura coccinea and 0.8 kb larger than that of Schisandra chinensis. The detected sequence length difference is predominantly attributable to the variation in the length of the intergenic spacer regions (Table 1).

Chloroplast genome map of Kadsura coccinea. Genes drawn outside of the circle are transcribed clockwise, while those inside are counterclockwise. Small single copy (SSC), large single copy (LSC), and inverted repeats (IRa, IRb) are indicated. The darker gray represents GC content in the inner circle, conversely the lighter one represents AT content.
All three Schisandraceae chloroplast genomes encoded 113 unique genes, including 79 protein-coding genes, 30 tRNA genes, and 4 ribosomal RNA genes. Of these, four protein-coding genes, four rRNA genes, and five tRNA genes were duplicated in the IR regions. There were 18 intron-containing genes (one class I intron in trnL-UAA and 17 class II introns), of which two genes, clpP and ycf3, contained two introns and the rest had only one intron each. In rps12, a trans-splicing event was observed, with the 5′ end located in the LSC region and the duplicated 3′ end in the IR region. The trnK-UUU gene harbours the largest intron, which contains the matK gene.
Repeat sequence analysis
Considering the role of chloroplast genome SSRs as important phylogenetic markers and valuable resources to indicate genome evolution, we screened and quantified six kinds of repeat patterns in Schisandraceae (Fig. 2). The total number of SSRs identified in the Kadsura coccinea, Schisandra chinensis, and Illicium oligandrum was 43, 74 and 100, respectively. The most abundant SSRs were A or T mononucleotide repeats, which accounted for approximately 60.5%, 56.8% and 69% of the total SSRs in Kadsura coccinea, Schisandra chinensis, and Illicium oligandrum, respectively, while the G or C repeats were rare. The number of penta- and hexanucleotides were slightly less than other repeats, such as di-, tri-, and tetranucleotides. Furthermore, the majority of SSRs of Kadsura coccinea, Schisandra chinensis, and Illicium oligandrum SSRs were located in LSC regions (83.7%, 78.3% and 74.0%, respectively), followed by SSC regions (11.6%, 16.2% and 16.0%, respectively) and IR regions (2.3%, 2.7% and 5.0%, respectively).

The distribution, type and presence of simple sequence repeats (SSRs) in the three chloroplast genome of Schisandraceae. (A) Number of different SSRs types. (B) Number of different SSRs in the LSC, SSC, and IR regions. (C) Number of identified SSR motifs in different repeat class types.
In addition to the SSRs, we employed REPuter and Tandem Repeats Finder to analyse the repeat sequences of the three chloroplast genomes (Fig. 3). The total number of repeats was 58 in Kadsura coccinea, 68 in Schisandra chinensis, and 61 in Illicium oligandrum. Kadsura coccinea contained 17 forward repeats, 16 palindrome repeats, and 25 tandem repeats. Schisandra chinensis contained 30 forward repeats, 13 palindrome repeats, and 25 tandem repeats, while Illicium oligandrum contained 8 forward repeats, 21 palindrome repeats, and 32 tandem repeats. Lengths of 30–40 repeats were the most common (average 56.7%), follow by 41–50 repeats. In addition, the proportions of repeats located in non-coding regions were higher than in coding regions.

Long repeat sequences in the three chloroplast genome of Schisandraceae. (A) Number of repeats. (B) Number of different repeats types. (C) Sequence length of repeats.
Sequence divergence and divergence hotspot
The mVISTA and DnaSP program were employed to analyse the overall sequence identity at the chloroplast genome level and to detect the divergent regions in the Schisandraceae chloroplast genome (Figs 4 and S2). The organization of the chloroplast genome among any of the compared genomes revealed a high degree of synteny and gene order conservation, suggesting an evolutionary conservation of these genomes at the genome-scale level. Overall, the results revealed higher divergence in non-coding regions than in coding regions. The coding regions with marked differences include the ycf1, accD and ndhF genes. The highest divergence in non-coding regions was found for rps16-trnQ, atpF-atpH, petN-psbM, trnT-psbC, ycf2-trnL, rpoB-trnC, ndhC-trnV, petA-psbJ, ndhF-rpl32, and rpl32-trnL. Notably, the LSC region and SSC region were more divergent than the IR regions.

Sequence comparison of the Illicium oligandrum, Kadsura coccinea and Schisandra chinensis chloroplast genomes generated by mVISTA. VISTA based similarity graphical information portraying sequence identity of Illicium oligandrum with reference A. indica chloroplast genomes. Grey arrows above the alignment indicate the orientation of genes. Purple bars represent exons, blue ones represent introns, and pink ones represent non-coding sequences (CNS). A cut-off of 50% identity was used for the plots. The Y-scale axis represents the percent identity within 50–100%. Dashed rectangles indicate highly divergent regions of Illicium oligandrum compared with Kadsura coccinea and Schisandra chinensis.
IR expansion and contraction
IR expansion and contraction often results in genome size variations among various plant lineages, which can be used to study the phylogenetic classification and the genome evolution among plant lineages13. In the present study, the IR boundary regions of three Schisandraceae species and three other early-diverging angiosperm species were compared, and the results showed that the border of the Schisandraceae chloroplast genome was slightly different from that of other genomes (Fig. 5). In Schisandraceae, the boundary was between trnL-CAA and ndhB on the IRb/LSC side and between ndhB and trnH-GUG on the IRa/LSC side. The boundary of IRb/LSC occurred between rps19 and rpl2 and between rpl2 and trnH-GUG on the IRa/LSC side, with 0 and 80 non-coding nucleotides between these two genes. The IR in Schisandraceae had a 10 kb contraction compared with other lineages.

Comparison of the border positions of the LSC, SSC, and IR regions among the six chloroplast genomes of basal angiosperms. Gene names are indicated in boxes, and their lengths in the corresponding regions are displayed above the boxes.
The IRa/SSC border extended into ycf1 resulting in a pseudogene in the three Schisandraceae chloroplast genomes that were compared. The length of the ycf1 pseudogene was 1,375 bp in Kadsura coccinea, 408 bp in Schisandra chinensis, and 403 bp in Illicium oligandrum. Furthermore, ndhF deviated from the IRb/SSC in Schisandra chinensis by 23 bp. The trnH-GUG gene was generally located downstream of the IRA/LSC border, and this gene is separated from the IRB/LSC border by 9 bp in Kadsura coccinea, 198 bp in Schisandra chinensis, and 480 bp in Illicium oligandrum. Overall, the IR boundary regions varied slightly in the Schisandraceae chloroplast genome.
Phylogenomic analysis
Chloroplast genome sequences have been widely used to reconstruct plant phylogenies42,43. To examine the phylogenetic position of Schisandraceae within angiosperms and the genus relationship within Schisandraceae, ML and BI methods of phylogenetic analysis were performed based on 82 protein-coding gene datasets from 66 plant taxa, including seven Schisandraceae species. The total alignment was 65,432 bp in length. Both the ML and BI trees had similar phylogenetic topologies, and most nodal support values were high (ML bootstrap support value >95/Bayesian posterior probability >0.99; Figs 6 and S1).

Phylogenetic tree reconstruction of 66 taxa using maximum likelihood and Bayesian inference based on concatenated sequences of 82 genes. ML topology shown with ML bootstrap support value/Bayesian posterior probability given at each node.
The trees provide support for the following relationships: Amborella and Nymphaeales are sisters to the remaining angiosperms; Schisandraceae is sister to a clade that includes magnoliids, monocots, and eudicots; magnoliids and monocots were both monophyletic; Amborellales, Nymphaeales, Austrobaileyales, and magnoliids formed the early-diverging angiosperms; basal eudicots were not monophyletic and Ceratophyllum was the sister to the remaining eudicots; rosids and asterids were each monophyletic; and Vitis and Paeonia were the earliest diverging lineage of rosids.
Schisandraceae was grouped in both ML and BI phylogenetic trees with 100% bootstrap values and 1.0 Bayesian posterior probability. Kadsura and Schisandra formed monophyly clades and were sisters to Illicium. Phylogenetic relationships among the five Illicium species were also established using this dataset.
Discussion
The organization of the Schisandraceae chloroplast genomes was similar to the angiosperm genome except the IR contraction. IR expansion/contraction also represents a highly variable region, which can be used to study molecular classification and the phylogenetic classification of plants. In this study, by comparing the inverted repeat/single copy (IR/SC) boundaries of the four basal angiosperms, we detected a 10 kb IR contraction in Schisandraceae. IR expansion/contraction has occurred multiple times in angiosperms based on the phylogenetic results. In the monocots, expansion of the IR has occurred on the IRa/LSC boundary resulting in a duplicate copy of the trnH-GUG gene next to rps19 at the IRb/LSC boundary. Adenophora stricta had a larger IR contraction with eight lost duplication genes compared with other Campanuloid species5. Small expansions and contractions of less than 1,000 bp was common in angiosperms, for example Oryza44. Three reasons may explain the diversification of IR boundary regions sequences. The first is intramolecular recombination, the second is the presence of multiple repeat sequences, and the third is the indels, which caused a mismatch that resulted in the upstream sequence becoming a single copy28.
SSRs are a type of 1–6 bp repeat frequently observed in chloroplast genomes, which can be used to unravel genome polymorphisms and perform population genetics of and across species45,46,47,48. In this study, the number of SSRs in the three Schisandraceae chloroplast genomes varied. Compared with the other species, the number of SSRs in the Kadsura coccinea was approximately the same as those in Forsythia suspense49 and Lagerstroemia50 but was two times lower than that in Illicium oligandrum. SSR primers for chloroplast genome are transferable across species and genera, because of the chloroplast genome conservation. The SSRs provided molecular markers for studying the genetic diversity and population structure of Schisandraceae species. Repeat sequences provided valuable resources to study genome recombination and rearrangement8. About sixty repeats in the Schisandraceae chloroplast genomes were found by REPuter, and this finding was similar in the plant51. Most repeat sequences and SSRs were distributed within noncoding regions, and chloroplast genome noncoding regions have been shown to be more variable than coding regions and to play an important role in phylogenetic studies in angiosperms52.
The chloroplast genomes are characterized by relatively small size, largely uniparental inheritance, conservation of gene content and order, and high copy number compared to the nuclear genome53. Hence, the chloroplast genome sequences have become widely used to resolve phylogenetic relationships among plants. With NGS technology, the chloroplast genome can be efficiently and economically obtained, and sequence data from the chloroplast genome have transformed plant systematics and contributed greatly to the current view of plant relationships54,55,56,57,58,59,60,61,62. A phylogenetic tree was constructed based on 82 protein-coding genes from 66 chloroplast genome sequences, which may represent the major angiosperm clades. ML and BI trees confirmed that Schisandraceae as one of the earliest diverging angiosperm lineages, and the position of the family was just internal to Amborella and Nymphaeales. The chloroplast genome data also established the internal relationship of Illicium with strong support. Therefore, the chloroplast genome sequence data were effective for inferring the backbone relationships among other family clades of angiosperms, as well as for resolving the phylogenetic relationship of species.
References
Daniell, H., Lin, C.-S., Yu, M. & Chang, W.-J. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biology 17, 1–29 (2016).
Dong, W., Xu, C., Cheng, T., Lin, K. & Zhou, S. Sequencing angiosperm plastid genomes made easy: A complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 5, 989–997 (2013).
Krause, K. Piecing together the puzzle of parasitic plant plastome evolution. Planta 234, 647–656 (2011).
Knox, E. B. The dynamic history of plastid genomes in the Campanulaceae sensu lato is unique among angiosperms. Proc Natl Acad Sci USA 111, 11097–11102 (2014).
Cheon, K.-S., Kim, K.-A. & Yoo, K.-O. The complete chloroplast genome sequences of three Adenophora species and comparative analysis with Campanuloid species (Campanulaceae). Plos One 12, e0183652 (2017).
Hong, C. P. et al. accD nuclear transfer of Platycodon grandiflorum and the plastid of early Campanulaceae. BMC Genomics 18, 607 (2017).
Guisinger, M. M., Kuehl, J. N. V., Boore, J. L. & Jansen, R. K. Genome-wide analyses of Geraniaceae plastid DNA reveal unprecedented patterns of increased nucleotide substitutions. Proc. Nat. Acad. Sci. USA 105, 18424–18429 (2008).
Weng, M.-L., Blazier, J. C., Govindu, M. & Jansen, R. K. Reconstruction of the Ancestral Plastid Genome in Geraniaceae Reveals a Correlation between Genome Rearrangements, Repeats, and Nucleotide Substitution Rates. Mol. Biol. Evol. 31, 645–659 (2014).
Marcussen, T. & Meseguer, A. S. Species-level phylogeny, fruit evolution and diversification history of Geranium (Geraniaceae). Mol. Phylogenet. Evol. 110, 134–149 (2017).
Schwarz, E. N. et al. Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids. J. Syst. Evol. 53, 458–468 (2015).
Wu, C. S., Wang, Y. N., Hsu, C. Y., Lin, C. P. & Chaw, S. M. Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 3, 1284–1295 (2011).
Wu, C. S. & Chaw, S. M. Highly rearranged and size-variable chloroplast genomes in conifers II clade (cupressophytes): evolution towards shorter intergenic spacers. Plant Biotechnol J, (2013).
Wang, R. J. et al. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8, 36 (2008).
Dong, W., Xu, C., Cheng, T. & Zhou, S. Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLOS ONE 8, e77965 (2013).
Borsch, T. & Quandt, D. Mutational dynamics and phylogenetic utility of noncoding chloroplast DNA. Plant Syst. Evol. 282, 169–199 (2009).
Ochoterena, H. Homology in coding and non-coding DNA sequences: a parsimony perspective. Plant Syst. Evol. 282, 151–168 (2009).
Sabir, J. et al. Evolutionary and biotechnology implications of plastid genome variation in the inverted-repeat-lacking clade of legumes. Plant Biotechnol J 12, 743–754 (2014).
Li, X. et al. Plant DNA barcoding: from gene to genome. Biological Reviews 90, 157–166 (2015).
Coissac, E., Hollingsworth, P. M., Lavergne, S. & Taberlet, P. From barcodes to genomes: extending the concept of DNA barcoding. Mol. Ecol. n/a-n/a (2016).
Perdereau, A., Klaas, M., Barth, S. & Hodkinson, T. R. Plastid genome sequencing reveals biogeographical structure and extensive population genetic variation in wild populations of Phalaris arundinacea L. in north-western Europe. GCB Bioenergy 9, 46–56 (2017).
Lima, M. S., Woods, L. C., Cartwright, M. W. & Smith, D. R. The (in)complete organelle genome: exploring the use and non-use of available technologies for characterizing mitochondrial and plastid chromosomes. Mol. Ecol. Resour. (2016).
Smith, A. C. families Illiciaceae and Schisandraceae. Sargentia; 7 (1947).
Wang, H., He, H.-J., Chen, J.-Q. & Lu, L. Palynological data on Illiciaceae and Schisandraceae confirm phylogenetic relationships within these two basally-branching angiosperm families. Flora - Morphology, Distribution, Functional Ecology of Plants 205, 221–228 (2010).
Zhang, J. et al. Evaluation of Four Commonly Used DNA Barcoding Loci for Chinese Medicinal Plants of the Family Schisandraceae. PLOS ONE 10, e0125574 (2015).
Youm, J. W., Han, S.-W., Seo, S. W., Lim, C. U. & Oh, S.-H. DNA barcoding of Schisandraceae in Korea. Korean J. Pl. Taxon 46, 273–282 (2016).
Luo, S. X., Chaw, S. M., Zhang, D. & Renner, S. S. Flower heating following anthesis and the evolution of gall midge pollination in Schisandraceae. Am. J. Bot. 97, 1220–1228 (2010).
Chase, M. W. & Reveal, J. L. A phylogenetic classification of the land plants to accompany APG III. Bot. J. Linn. Soc. 161, 122–127 (2009).
Hansen, D. R. et al. Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae). Mol. Phylogenet. Evol. 45, 547–563 (2007).
Guo, H. et al. Complete chloroplast genome sequences of Schisandra chinensis: genome structure, comparative analysis, and phylogenetic relationship of basal angiosperms. Sci China Life Sci, (2017).
Li, J., Wang, S., Jing, Y., Wang, L. & Zhou, S. A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot. 48, 72–78 (2013).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455–477 (2012).
Li, B. et al. Development of chloroplast genomic resources for Akebia quinata (Lardizabalaceae). Conservation Genetics Resources 8, 447–449 (2016).
Huang, D. I. & Cronk, Q. C. B. Plann: A Command-Line Application for Annotating Plastome Sequences. Applications in Plant Sciences 3, 1500026 (2015).
Conant, G. C. & Wolfe, K. H. GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics 24, 861–862 (2008).
Kurtz, S. et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642 (2001).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279 (2004).
Librado, P. & Rozas, J. DnaSPv5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Sun, L. et al. Chloroplast phylogenomic inference of green algae relationships. Sci. Rep. 6, 20528 (2016).
Goremykin, V. V., Nikiforova, S. V., Cavalieri, D., Pindo, M. & Lockhart, P. The Root of Flowering Plants and Total Evidence. Syst. Biol. 64, 879–891 (2015).
Asaf, S. et al. The Complete Chloroplast Genome of Wild Rice (Oryza minuta) and Its Comparison to Related Species. Frontiers in Plant Science 8 (2017).
Zhou, S. et al. How many species of bracken (Pteridium) are there? Assessing the Chinese brackens using molecular evidence. Taxon 63, 509–521 (2014).
Qi, W. et al. High-throughput development of simple sequence repeat markers for genetic diversity research in Crambe abyssinica. BMC Plant Biol. 16, 139 (2016).
Yu, J. et al. PMDBase: a database for studying microsatellite DNA and marker development in plants. Nucleic Acids Res. 45, D1046–D1053 (2017).
Ebert, D. & Peakall, R. Chloroplast simple sequence repeats (cpSSRs): technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 9, 673–690 (2009).
Wang, W. et al. The Complete Chloroplast Genome Sequences of the Medicinal Plant Forsythia suspensa (Oleaceae). Int. J. Mol. Sci. 18, 2288 (2017).
Xu, C. et al. Comparative Analysis of Six Lagerstroemia Complete Chloroplast Genomes. Front Plant Sci 8, 15 (2017).
Dong, W. et al. Phylogenetic Resolution in Juglans Based on Complete Chloroplast Genomes and Nuclear DNA Sequences. Front Plant Sci 8, 1148 (2017).
Dong, W., Liu, J., Yu, J., Wang, L. & Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. Plos One 7, e35071 (2012).
Yuan, Z. et al. The pomegranate (Punica granatum L.) genome provides insights into fruit quality and ovule developmental biology. Plant Biotechnol J 0 (2018).
Ruhfel, B. R., Gitzendanner, M. A., Soltis, P. S., Soltis, D. E. & Burleigh, J. G. From algae to angiosperms-inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol. Biol. 14, 23 (2014).
Barrett, C. F., Davis, J. I., Leebens-Mack, J., Conran, J. G. & Stevenson, D. W. Plastid genomes and deep relationships among the commelinid monocot angiosperms. Cladistics 29, 65–87 (2013).
Ross, T. G. et al. Plastid phylogenomics and molecular evolution of Alismatales. Cladistics 32, 160–178 (2016).
Carbonell-Caballero, J. et al. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus. Citrus. Mol. Biol. Evol. 32, 2015–2035 (2015).
Zhong, B. et al. Streptophyte algae and the origin of land plants revisited using heterogeneous models with three new algal chloroplast genomes. Mol. Biol. Evol. 31, 177–183 (2014).
Jansen, R. K. et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Nat. Acad. Sci. USA 104, 19369–19374 (2007).
Ma, P. F., Zhang, Y. X., Zeng, C. X., Guo, Z. H. & Li, D. Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 63, 933–950 (2014).
Givnish, T. J. et al. Phylogenomics and historical biogeography of the monocot order Liliales: out of Australia and through Antarctica. Cladistics 32, 581–605 (2016).
Williams, A. V., Miller, J. T., Small, I., Nevill, P. G. & Boykin, L. M. Integration of complete chloroplast genome sequences with small amplicon datasets improves phylogenetic resolution in Acacia. Mol. Phylogenet. Evol. 96, 1–8 (2016).
Acknowledgements
This work was supported by the National Forest Genetic Resources Platform 2017.
Author information
Authors and Affiliations
Contributions
B.L. and Y.Z. designed the experiment; B.L. collected samples, performed the experiment, analyzed the data and wrote the manuscript; All of the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, B., Zheng, Y. Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Sci Rep 8, 9285 (2018). https://doi.org/10.1038/s41598-018-27453-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27453-7
This article is cited by
-
Plastid genome data provide new insights into the dynamic evolution of the tribe Ampelopsideae (Vitaceae)
BMC Genomics (2024)
-
Chloroplast genomes of Caragana tibetica and Caragana turkestanica: structures and comparative analysis
BMC Plant Biology (2024)
-
The complete Chloroplast genome of Stachys geobombycis and comparative analysis with related Stachys species
Scientific Reports (2024)
-
Regardless of having identical photosynthetic pathways, chloroplast genomes vary depending on whether the host plant is monocotyledonous or dicotyledonous
Genetic Resources and Crop Evolution (2024)
-
Complete chloroplast genome and phylogenetic analysis of Anemone shikokiana
Molecular Biology Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
