
Abstract
Influenza virus (IV) infections cause severe respiratory illnesses that can be complicated by bacterial super-infections. Previously, we identified the cellular Raf-MEK-ERK cascade as a promising antiviral target. Inhibitors of MEK, such as CI-1040, showed potent antiviral activity. However, it remained unclear if this inhibitor and its active form, ATR-002, might sensitize host cells to either IV or secondary bacterial infections. To address these questions, we studied the anti-pathogen activity of ATR-002 in comparison to CI-1040, particularly, its impact on Staphylococcus aureus (S. aureus), which is a major cause of IV super-infections. We analysed IV and S. aureus titres in vitro during super-infection in the presence and absence of the drugs and characterized the direct impact of ATR-002 on bacterial growth and phenotypic changes. Importantly, neither CI-1040 nor ATR-002 treatment led to increased bacterial titres during super-infection, indicating that the drug does not sensitize cells for bacterial infection. In contrast, we rather observed reduced bacterial titres in presence of ATR-002. Surprisingly, ATR-002 also led to reduced bacterial growth in suspension cultures, reduced stress- and antibiotic tolerance without resistance induction. Our data identified for the first time that a particular MEK-inhibitor metabolite exhibits direct antibacterial activity, which is likely due to interference with the bacterial PknB kinase/Stp phosphatase signalling system.
Similar content being viewed by others

Introduction
Influenza viruses cause infections of the respiratory tract resulting in severe diseases, especially in high-risk patients. Fast illness progression and high mortality rates are often associated with secondary bacterial infections induced by common colonizers of the nasopharynx, such as S. aureus or Streptococcus pneumoniae (S. pneumoniae)1,2,3,4,5. In contrast to infections with streptococci that occur at late phases following viral clearance, S. aureus is mostly detected during concomitant IV infections. Furthermore, S. aureus gives rise to a continuously growing challenge in the clinics due to resistance development, such as Methicillin-resistant S. aureus (MRSA) strains6,7,8. Similarly, resistant IV variants evolve and vaccine evasion occurs regularly. Thus, there is an urgent need for new therapeutics against both pathogens9,10,11,12,13.
Novel antiviral strategies to combat influenza are based on the fact that IV, as intracellular pathogens, strongly depend on the cellular signalling machinery14. Thus, cellular virus-supportive functions are promising candidates for alternative approaches thereby reducing the likelihood to provoke viral resistance. In contrast, S. aureus division has been mostly thought of as host-cell independent. By directing novel antibacterial treatments towards inhibition of bacterial virulence factors expressed during infection, these compounds may also exhibit a lower potential to induce resistance. Additionally, there is accumulating evidence that S. aureus also uses cellular signalling for its own benefits during infection15, but such bacterial-supportive cellular factors have yet to be characterized.
The cellular Raf-MEK-ERK pathway is involved in the nuclear export of newly synthesized viral ribonucleoproteins (vRNPs) during IV replication. Prior studies have shown that compounds inhibiting this pathway exhibit significant anti-influenza activity in vitro and in vivo16,17. The inhibition of this pathway by use of specific MEK-inhibitors like U0126 and CI-1040 resulted in a reduction of viral titres without induction of resistances11,16,17,18,19. Especially CI-1040 was shown to be a potent compound for anti-influenza intervention with a prolonged treatment window when compared to standard of care18. In vivo CI-1040 is metabolized into its acidic form ATR-002 (originally termed PD0184264), which is the major initial metabolite after oral administration and appears to be the biological active entity20,21. Yet, the anti-pathogen activity of ATR-002 has not previously been tested.
One concern of using cellular signalling inhibitors is sensitization of a flu-infected host to subsequent super-infections. In fact, the activation of the Raf-MEK-ERK pathway and downstream signalling cascades has been demonstrated upon bacterial infections22,23 and Raf-MEK-ERK-mediated signalling appears to be important in immune responses during singular IV and bacterial super-infection with S. aureus and S. pneumoniae23,24.
It was assumed for a long time that serine/threonine (Ser/Thr) kinases, like MEK and ERK, are exclusively expressed by eukaryotes, but recently eukaryotic-like Ser/Thr kinases have been identified in the majority of bacterial families25. These prokaryotic kinases are involved in various cellular functions, like metabolic processes, cell wall metabolism, environmental responses and pathogenicity26,27,28. Notably, S. aureus expresses the Ser/Thr kinase PknB, that shares high homology with cellular MAPK29. Besides metabolic processes, PknB is involved in regulation of bacterial antibiotic susceptibility and other pathogenicity determining processes including stress response and growth behaviour30,31,32,33,34.
One aim of the present study was to elucidate whether MEK-inhibitors, such as CI-1040 and its metabolite ATR-002, would enhance replication of bacteria during IV/S. aureus super-infection. Furthermore, we aimed to explore the anti-pathogen activity of ATR-002, especially the direct impact on bacterial growth.
Results
Treatment with CI-1040 or ATR-002 does not sensitize cells for secondary bacterial infections
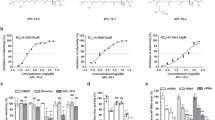
IV infection results in enhanced expression of antiviral cytokines, most importantly type I IFNs, that activate critical downstream antiviral responses and may also potentiate subsequent bacterial infections2,35,36,37. Since the Raf-MEK-ERK pathway is involved in expression of some of these cytokines we wondered whether treatment with CI-1040 or ATR-002 would sensitize cells for a secondary S. aureus infection. Cell cultures of immortalized human alveolar basal epithelial cells (A549) were infected with IV and S. aureus in the presence or absence of the inhibitors. Inhibitors were used at 10 µM, which is considered to be a concentration still specific for MEK inhibition in cell culture38. Microscopic examination revealed that super-infection with both pathogens resulted in a highly increased cytopathic effect (CPE) compared to singular infections (Figure S1, upper panel). The CPE was completely abolished in presence of ATR-002 (Figure S1, lower panel) indicating reduced viral replication. Similar results were obtained upon MEK inhibition where both CI-1040 and ATR-002 reduced IV titres, with a less pronounced effect of ATR-002 (Fig. 1a,b). Due to variations between experiments, the effects of CI-1040 and ATR-002 on viral replication may not be strong, nevertheless, the effects seen were reproducible. Importantly, treatment with CI-1040 did not sensitize cells for a secondary infection with S. aureus as no changes in intracellular bacterial load could be detected (Fig. 1d). Surprisingly, administration of ATR-002 even resulted in reduced intracellular bacterial titres (Fig. 1e). Comparable results were obtained when CI-1040 or ATR-002 were administered at later times during on-going infection (Fig. 1c,f). To rule out that the reduction in viral and intracellular bacterial replication was a result of a cytotoxic effect of ATR-002 on A549 cells, cell viability in presence of increasing concentrations was monitored for 24 and 48 hours. Additionally, a LDH-Assay was performed to determine membrane rupture due to inhibitor treatment (Figure S2). Furthermore, we excluded a potential impact of ATR-002 on bacterial infectivity by performance of bacterial internalization assays in presence or absence of the inhibitor (Figure S3).

ATR-002, but not CI-1040, decreases intracellular bacterial titres. A549 cells were pre-treated (a,b,d,e) with 10 µM CI-1040, ATR-002 or solvent (DMSO) for 60 min and then infected with IV (H7N7) at a MOI of 0.001 at 37 °C. Alternatively, cells were left untreated (c,f) and infected with IV (H1N1) at a MOI of 0.01 at 37 °C. After 30 min the virus dilution was removed, cells were rinsed with PBS and supplemented with invasion medium with or without S. aureus 6850 (6850) (MOI 0.1) in the presence of 10 µM CI-1040, ATR-002 or solvent control. 3 h post bacterial infection cells were treated with lysostaphin (2 µg/mL) for 20 min to remove extracellular bacteria. Cells were then washed and supplemented with infection medium containing the inhibitor or solvent. After a total incubation period of 24 h (post viral infection) viral (a–c) and intracellular bacterial titres (d–f) were analysed. Results represent means + SD of three individual experiments. Statistical significance was evaluated by one-way ANOVA followed by Tukey’s multiple comparisons test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
S. aureus growth in suspension cultures is strongly inhibited by ATR-002, but only slightly affected by other MEK-inhibitors
Upon administration of ATR-002 intracellular S. aureus titres were slightly reduced. While S. aureus does not rely on internalization into cells, bacteria express kinases homologous to eukaryotic MAPK. Consequently, we examined whether the reduced bacterial titres were an indirect result of the inhibited Raf-MEK-ERK pathway or due to direct interaction of ATR-002 with the bacteria. To assess this, we analysed the impact of increasing concentrations of ATR-002 and other MEK-inhibitors (U0126 and CI-1040) on suspension cultures of the Methicillin-sensitive S. aureus strain 6850 (MSSA) or the MRSA strain USA300 and calculated viable bacteria. Surprisingly, the presence of ATR-002 provoked a strong reduction of bacterial growth of the MSSA and to a slightly lower extent of MRSA strain (Fig. 2a,b). This growth retention by ATR-002 was already detectable shortly after inoculation of bacterial cultures and by far exceeded the effects of the parental compound CI-1040 (Fig. 2c). During over-night treatment with even higher concentrations of CI-1040 no comparably strong growth inhibition was observed. Although a significant reduction of S. aureus growth could be detected using 50 µM of U0126, this growth retention was considerably low compared to growth inhibition caused by ATR-002 (Fig. 2d,e). To confirm a medium-independent effect of ATR-002 on bacteria, we repeated these experiments in different media (Figure S4).

S. aureus growth is strongly impaired exclusively in presence of ATR-002. Over-day cultures of S. aureus 6850 or the MRSA strain USA300 were set to 20 CFU/ml and treated with different MEK-inhibitors (as indicated) over-night at 37 °C and 5% CO2. The OD600 was measured and remaining cultures were washed once with PBS. Bacterial titres were determined by serial dilutions on BHI agar plates (a,b,d,e). Growth curves were generated by dilution of 25 µl of untreated over-night cultures in 8 ml of fresh medium and were incubated over a time course of 6 h in presence of 50 µM CI-1040, ATR-002 or DMSO. The OD600 was measured at the indicated time points (c). Results represent means + SD (a,b,d,e); +/− SD (c) of three independent experiments. Statistical significance was analysed by one-way ANOVA followed by Dunnett’s multiple comparisons test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
ATR-002 exhibits bacteriostatic properties without induction of resistance in vitro
To further classify the antibacterial action of ATR-002, growth behaviour of S. aureus was monitored in comparison to a known bactericidal agent, gentamicin. Treatment of MSSA and MRSA with 20 µM of ATR-002 led to a stronger reduction in bacterial titres than 0.5 µg/ml of gentamicin, but was less pronounced than treatment with 1.5 µg/ml of gentamicin (Fig. 3a,b).

ATR-002 exhibits a low bacteriostatic effect without resistance induction. Over-day cultures of S. aureus 6850 or the MRSA strain USA300 were set to 20 CFU/ml and treated as indicated over-night at 37 °C and 5% CO2. The OD600 was measured, cultures were washed once with PBS and viable bacteria were counted by serial dilutions on BHI agar plates (a,b). To classify the antibacterial action, a time-of-addition assay was performed (c). Therefore, sub-cultures of an over-night culture of S. aureus 6850 were prepared and were treated for 9 h as indicated. At 0, 3, 6 and 9 h the OD600 was measured and titres were calculated. Additionally, long-term treatment with either ATR-002, Gentamicin or Erythromycin was performed to test for resistance development (d). Cultures were grown for 24 h in the presence or absence of the substances, the OD600 was measured and then cultures were set to 100 CFU/ml and grown again for 24 h. This procedure was repeated for 17 days. Additional results of testing for cross-resistance development are summarized in Table S1. Data represent means + SD of three independent experiments. Statistical significance was analysed by one-way ANOVA followed by Dunnett’s multiple comparisons test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Additionally, a time-of-addition assay was performed to investigate the action of ATR-002 on bacterial multiplication over time. Therefore, U0126, ATR-002 or gentamicin were added to S. aureus cultures. Bacterial amounts were analysed over a time course of 9 h starting at the time of compound addition. Interestingly, in the presence of ATR-002 inhibition of bacterial growth was even higher than caused by 2 µg/ml of gentamicin at early times of incubation (Fig. 3c). To shed some first light on the mode of action the bacterial cultures were diluted after 9 h in BHI medium and were further grown without substance addition. All cultures reached the turbidity of the untreated samples indicating a bacteriostatic rather than a bacteriocidal effect of ATR-002.
The frequent emergence of antibiotic resistant strains represents a major problem in the clinics. To test whether ATR-002 provokes resistance development, cultures were constantly treated for 17 days in presence of ATR-002, gentamicin or erythromycin. Resistance development to gentamicin occurred during the first week of treatment. In contrast, treatment with ATR-002 did not induce resistance similar to treatment with the macrolide erythromycin (Fig. 3d). Additionally, constant treatment with ATR-002 did not induce cross-resistances as shown by unchanged or even reduced MIC concentrations calculated for a variety of antibiotics after ATR-002 treatment (Table S1).
ATR-002 treatment results in reduced antibiotic and stress resistance of S. aureus
Since ATR-002 is a cellular kinase inhibitor, bacterial Ser/Thr kinases, like PknB of S. aureus, would be a likely target candidate. S. aureus lacking PknB shows reduced growth, which correlates with increased antibiotic sensitivity and reduced stress tolerance32,39. Thus, we addressed the impact of ATR-002 on antibiotic susceptibility and heat tolerance of S. aureus. Therefore, we determined the MIC against different antibiotics after over-night treatment. Surprisingly, treatment with ATR-002 increased the susceptibility of both MSSA (S. aureus 6850) and MRSA (USA300) strains against various antibiotics, in particular penicillin susceptibility by up to tenfold. Here, representative data are shown for MSSA (Fig. 4a, Tables S2 and S3). Next, the ability of S. aureus to grow under stress conditions was evaluated after treatment. Therefore, the differentially treated cultures were set to an OD of 1, diluted in BHI and further incubated at 42 °C to induce heat stress. Compared to previously solvent-treated cultures, ATR-002-treated bacteria were more sensitive towards heat stress. Consequently, MSSA and MRSA strains showed a delayed growth (Fig. 4b). Heat stress response of these strains was further evaluated after treatment with ATR-002 in different growth media (Figure S5).

Treatment with ATR-002 results in increased antibiotic susceptibility and reduced stress resistance of S. aureus. Cultures of S. aureus 6850 (20 CFU/ml), originated from over-day cultures, were prepared in BHI medium and treated as indicated over-night at 37 °C. Subsequently, bacterial cultures were washed with PBS and resuspended in 1 ml PBS. MICs were determined using 100 µl of over-night cultures and a M.I.C.Evaluator strip on agar plates. After 24 h at 37 °C plates were analysed and the concentration preventing growth was termed as MIC (a). A summary of three independent experiments is shown in Table S2. Solvent or inhibitor treated over-night cultures were further cultivated after dilution in BHI medium at 42 °C to induce heat stress for 6 h. Then, bacterial counts were analysed (b). Data represent three individual experiments. Statistical significance was analysed by one-way ANOVA followed by Dunnett’s multiple comparisons test (**p < 0.01; ***p < 0.001).
The antibacterial action of ATR-002 is mediated by interaction with the PknB/Stp signalling system of S. aureus
ATR-002 clearly exhibits antibacterial properties, correlating with reduced antibiotic and stress resistance. S. aureus strains lacking PknB share this phenotype39,40,41,42. Hence, we addressed whether PknB is involved in the antibacterial action of ATR-002. Therefore, we performed an in vitro kinase assay to test for a direct interaction with the purified kinase domain (PknB1–291) (generated and provided by the Ohlsen laboratory, Würzburg). The activity of PknB is regulated via autophosphorylation. Thus, we assessed ATR-002 action on PknB autophosphorylation as well as target phosphorylation of MBP (Fig. 5a). In the absence of ATP neither PknB nor MBP were phosphorylated, however, the addition of ATP induced both autophosphorylation of PknB and phosphorylation of MBP. Surprisingly, treatment with ATR-002 did not reduce phosphorylation, indicating that in this system, the inhibitor was not able to block activity of the kinase domain. However, here the influence of the C-terminal domain of PknB or the impact of other bacterial factors that may affect kinase activity during bacterial growth cannot be measured. Therefore, we performed an over-night treatment of different wildtype (WT) S. aureus strains and respective deletion mutants lacking either the kinase (ΔpknB), the corresponding phosphatase (Stp) (Δstp) or both enzymes (ΔpknB/stp) with ATR-002 (Fig. 5b–e). ATR-002 treatment clearly reduced bacterial growth of WT strains. Surprisingly, the ΔpknB and the Δstp strains were still sensitive to ATR-002. In contrast to that, the double knock-out strains completely lost sensitivity to the inhibitor. These data indicate, that neither PknB nor Stp alone seem to be the targets for the antibacterial action of ATR-002. The inhibitor rather seems to interfere with the complex interplay of both enzymes suggesting that ATR-002 leads to a disturbance of this signalling system.

The antibacterial action of ATR-002 is mediated via interaction with signalling pathways of S. aureus. The impact of ATR-002 on PknB activity was analysed by an in vitro kinase assay (a). Therefore, 0.5 µg of the purified kinase domain (PknB1–291) were incubated in kinase buffer in the presence or absence of 1 µl ATP (2 mM) and 8 µCi 32P-ATP and increasing amounts of the MEK-inhibitor ATR-002 (as indicated) for 1 h at 37 °C. The reaction was stopped by addition of 5 × SDS sample buffer. Following SDS page and Western Blot, the phosphorylation of PknB and MBP was analysed by radiography. Additionally, wildtype (WT) or mutant strains lacking either a functional kinase (ΔpknB), the phosphatase (Δstp) or both (ΔpknB/stp) were treated over-night in the presence of 20 µM ATR-002 or solvent (DMSO) at 37 °C. Afterwards, bacterial titres were calculated by plating serial dilutions on BHI agar plates (b–e). Data represent one representative blot of three experiments (a) or means + SD of three independent experiments (b–e). Western Blot data show cropped blots from two full-length blots, which are provided in Figure S8. Statistical significance was analysed by one-way ANOVA followed by Dunnett’s multiple comparisons test (*p < 0.05, **p < 0.01; ***p < 0.001; ****p < 0.0001).
The impact of ATR-002 on bacterial growth is not restricted to S. aureus
The presence of bacterial Ser/Thr kinases is described to be conserved among different bacterial genera25,26,43. Therefore, we tested whether ATR-002 has antibacterial properties to other bacteria. Hence, the impact on Escherichia coli (E. coli), Bacillus subtilis (B. subtilis) and Mycobacterium abscessus (M. abscessus) was analysed. While growth of E. coli and M. abscessus was not affected (not shown), a strong decrease in viable bacterial counts was observed for B. subtilis in presence of 10 µM ATR-002, and was completely abolished with higher concentrations (Fig. 6a). Besides S. aureus, one of the most abundant bacteria to cause bacterial super-infections is S. pneumoniae44,45,46. Thus, activity of ATR-002 against S. pneumoniae strains D39 and TIGR4 was tested. In the presence of 20 µM ATR-002 a reduction in growth of both strains was observed (Fig. 6b) indicating a broader antibacterial action of ATR-002.

The inhibitory effect of ATR-002 is not restricted to S. aureus. To test for a potential antibacterial effect on other bacterial species, over-night cultures of B. subtilis (a) and the S. pneumoniae wild-type strains D39 and TIGR4 (b) were incubated with either solvent or different concentrations of the MEK-inhibitor ATR-002 (as indicated) for 18 h. Afterwards, bacterial load was determined via measurement of the OD600 and plating of serial dilutions on BHI agar or columbia blood agar plates, respectively. Data represent means + SD of three independent experiments. Statistical significance was analysed by one-way ANOVA followed by Dunnett’s multiple comparisons test (**p < 0.01; ***p < 0.001; ****p < 0.0001).
Discussion
Bacterial super-infections following influenza infections represent an important public health problem and occur during both influenza epidemics and pandemics. Unfortunately, only a limited arsenal of potent anti-infectives against IV and/or S. aureus is available and drug-resistance is a major concern. For anti-influenza intervention, the use of inhibitors targeting cellular pathways came into the focus of research. Previous studies have shown that blockade of the Raf-MEK-ERK pathway leads to reduced progeny virus production in vitro and in vivo with a high barrier towards viral resistance16,17,18. However, it remained unclear whether inhibition of this pathway in primary IV infection could predispose the host to subsequent bacterial infections.
While we could not only rule out such a sensitization for bacteria, our findings rather indicate the opposite effect. We show that the metabolic conversion of CI-1040 into its metabolite ATR-002 turns the MEK-inhibitor into an antibacterial compound. This not only resulted in reduced intracellular bacterial titres of S. aureus in a super-infection scenario but also in strongly reduced bacterial growth in suspension cultures.
The efficacy of cellular MEK-inhibitors against IV infections has been shown in several studies by us and others16,17,18. However, in case of secondary bacterial infections, the impact of MEK-inhibitor treatment has not been analysed so far. We show, that administration of ATR-002 during super-infection of IV and S. aureus leads to reduced viral titres and inhibition of the CPE. Additionally, intracellular bacterial titres were slightly reduced upon treatment, which could only be observed with ATR-002 and not with CI-1040. Still, these differences in pathogen load do not fully explain the great reduction of the induced CPE. Upon infection, bacteria secrete virulence factors such as toxins, which in turn can lead to tissue damage36,47,48. Furthermore, intracellular bacteria can cause cell death upon phagosomal escape47,49,50. It cannot be ruled out that administration of ATR-002 might interfere with secretion of bacterial virulence factors and hence, lead to a reduced destruction of surrounding cells and tissue47,48. Beside a rather low reduction of intracellular bacterial load, we observed a strong inhibition of bacterial growth in suspension cultures upon ATR-002 treatment correlating with decreased antibiotic and stress resistance. This might be an exclusive function of ATR-002 rather than a general feature of MEK-inhibitors as this strong impairment in growth was not visible with other MEK-inhibitors. The fact that the parental compound CI-1040 did not show any antibacterial effect indicates that metabolic conversion to ATR-002 leads to differences in chemical structure and thereby to higher binding affinity and accessibility to the bacterial target(s). Recently published data show the impact on bacterial growth and pathogenicity in presence of specific inhibitors against the bacterial kinase PknB41. Notably, a retention in bacterial growth accompanied by increased antibiotic susceptibility was observed, when bacteria were incubated with a specific Stk1 inhibitor41. Furthermore, the dual administration of kinase inhibitors in combination with antibiotics resulted in increased bactericidal properties of ceftriaxone and cefotaxime41. Comparable data were also published by Vornhagen et al. showing increased antibiotic sensitivity upon inhibitor treatment in case of Methicillin-resistant strains39. Our data strongly support the hypothesis that the inhibitory action of ATR-002 is at least partially mediated by interaction with the bacterial PknB-Stp signalling system. It is likely that more than one bacterial target of this system is involved in the observed phenotype. For example, a crosstalk of the PknB-Stp pathway with the two-component system WalKR was demonstrated. This regulator is greatly involved in cell wall synthesis and virulence of S. aureus and might represent an additional target of ATR-00251. Beside the involvement of other pathways, mutations of S. aureus PknB were shown to affect bacterial cell wall structure52. At this point we cannot rule out, if changes in cell wall structure and thickness in bacteria harbouring the double knock-out of PknB and Stp also play a role in the observed phenotype. This might explain the lack of antibacterial activity of ATR-002 in gram-negatives as in these bacteria cell wall structure is completely different to gram-positives like S. aureus. While the direct target(s) of ATR-002 may not be clear, interference with a more complex system rather than one particular enzyme would have a major advantage: The likeliness of resistance development may be greatly reduced, as it has been observed in our assays. While the antibacterial action of ATR-002 is intriguing, it has to be pointed out that the data provided so far are limited to in vitro experiments. The potential of a future therapeutic use needs to be further explored in in vivo models, especially in an IV and S. aureus super-infection model. Another issue that needs final experimental clarification is, whether conversion of CI-1040 to ATR-002 changes its target profile on cellular kinases and pathways. Due to the fact that we do not know the final target of the MEK pathway on viral structures in cells, and crosstalk of the Raf pathway with other signalling cascades has been described53, it cannot completely be ruled out that the intracellular anti-pathogen action of ATR-002 involves still other pathways or kinases. Recently we showed that the inhibitor Vemurafenib, that blocks the MEK activator Raf acts antiviral via interference with multiple pathways54. While this may serve as an example of crosstalk, it has to be pointed out that Vemurafenib works via a completely different mode of action compared to ATR-002. Furthermore, MEK inhibitors are described to be very specific to their target kinase. Finally, such an intracellular crosstalk would be irrelevant for the direct antibacterial action of ATR-002.
In summary, we show for the first time that metabolic conversion of the cellular MEK-inhibitor CI-1040 into ATR-002 generates an antibacterial compound. Beside interference with the influenza life cycle by interrupting the virus-supportive Raf-MEK-ERK pathway, ATR-002 strongly decreases growth of S. aureus and other bacterial species. Our findings may have important implications for the development of strategies to treat post-influenza super-infections or infections with antibiotic-resistant bacteria.
Methods
Cell lines, viral and bacterial strains
The human alveolar basal lung epithelial cell line (A549) was cultivated in Dulbecco’s Modified Eagle Medium (DMEM) and Madin-Darby canine kidney cells (MDCK) were cultivated in minimal essential medium. Both media were supplemented with 10% fetal bovine serum (FBS).
All viral and bacterial strains used in the present study are listed in Table S4.
Viral stocks were generated by passaging of the viruses on MDCK cells. Infectious particles were calculated via standard plaque assay. Bacteria were stored as glycerol stocks [comprised of brain heart infusion (BHI) and 30% glycerol] at −80 °C and on blood agar plates until further use.
Super-infection of A549 cells with IV and S. aureus
An over-night culture of S. aureus was prepared prior to infection. A single colony was selected and inoculated in 5 ml BHI medium for 16–18 h. Confluent A549 cells were prepared by seeding 6- or 12-well plates (5 × 105 or 2.5 × 105 cells/well) 24 h before the experiment. For viral infection, cells were washed with phosphate-buffered saline (PBS) and infected with 500 µl of the virus stock adjusted in PBS/INF [0.2% bovine serum albumin (BSA), 1 mM MgCl2, 0.9 mM CaCl2, 100 U/ml penicillin, 0.1 mg/ml streptomycin] to the desired multiplicity of infection (MOI). After 30 min, cells were washed with PBS and the secondary bacterial infection was performed. For this, the indicated MOI was adjusted in 1 ml DMEM/INV [1% human serum albumin, 25 nmol/l HEPES] and cells were incubated for 3 h. To avoid bacterial over-growth, extracellular bacteria were removed by an antibiotic wash step. Therefore, cells were washed with PBS and incubated with 1 ml of DMEM/FBS [10% FBS, 2 μg/ml lysostaphin] for 20 min. Subsequently, cells were washed with PBS and incubated in 1 ml of DMEM/BA [0.2% BSA, 1 mM MgCl2, 0.9 mM CaCl2] until the end of the experiment. For experiments extending one influenza replication cycle (>8 h) Trypsin solo (2 µg/ml) was added during over-night incubation to allow efficient infection over time. All incubation steps were performed at 37 °C, 5% CO2. The infection scheme is summarized in Figure S6.
Inhibitor treatment during super-infection
The MEK-inhibitors U0126 (Taros Chemicals GmbH & Co. KG), CI-1040 and ATR-002 (originally PD0184264) (kindly supplied by ATRIVA Therapeutics GmbH) were dissolved in DMSO to prepare 10 mM stocks. Before infection, cells were washed with PBS and incubated with 1 ml medium containing the inhibitor (as indicated) or DMSO for 1 h at 37 °C, 5% CO2. Afterwards, cells were infected with IV and S. aureus as described above. During incubation with DMEM/INV and DMEM/BA, media were supplemented with the inhibitors. All inhibitors are described in Table S5. Chemical structures of CI-1040 and the metabolite ATR-002 are displayed in Figure S7.
Cytotoxicity testing
Potential cytotoxic effects of ATR-002 on A549 cells were determined by counting of viable cells compared to DMSO-treated cells upon 24 and 48 h. Medium containing either DMSO or increasing amounts of ATR-002 was added to A549 cells seeded in 6-well plates and were incubated for 24 or 48 hours. After the respective times viable cells were counted by staining with trypan blue, which selectively stains non-viable cells due to a permissive cell wall. Additionally, supernatants were taken for measurement of LDH release due to membrane rupture. For this, the CytoSelect LDH Cytotoxicity Assay Kit (CBA 241) was used according to manufacturer’s instructions. Values were normalized to DMSO-treated cells and are shown as % viability.
Calculation of viral and intracellular bacterial titres
For determination of viral replication during the course of infection, the number of infectious particles in the supernatants was measured as described previously55. Intracellular bacterial titres were determined by washing infected cells twice with PBS and lysis with 2 ml of ddH2O at 37 °C, 5% CO2 for 30 min. Afterwards, lysates were transferred into tubes and centrifuged at 4000 rpm, 4 °C for 10 min. Pellets were resuspended in 1 ml PBS, serially diluted and incubated for 24 h at 37 °C on agar plates. Bacterial titres were calculated as colony forming units (CFU)/ml.
Inhibitor treatment during bacterial growth
U0126, CI-1040 and ATR-002 were added to suspension cultures of S. aureus, B. subtilis or S. pneumoniae. Cultures were prepared by inoculating 5 ml BHI medium with a single bacterial colony and incubating at 37 °C, 5% CO2 for 8 h. Subsequently, cultures were centrifuged at 4000 rpm, 4 °C for 5 min and the pellet was washed with PBS. Afterwards, the bacterial suspension was adjusted to an optical density (OD600) of 1 in PBS, which represented a viable bacterial count of 5 × 108 CFU/ml in case of S. aureus (confirmed by plating). For over-night treatment, 5 ml BHI medium were inoculated with 20 CFU/ml and incubated for 16 h in the presence of DMSO, inhibitor (as indicated) or medium at 37 °C, 5% CO2. Thereafter, the OD600 was measured and cultures were washed with PBS. Bacterial titres were performed by resuspending the pellet in 1 ml PBS and serial dilutions on BHI agar plates. Additionally, growth inhibition was tested in a chemically defined medium (described previously by Schoenfelder et al.56, RPMI cell culture medium as well as in Mueller-Hinton II medium (Table S5).
Stress tolerance of S. aureus after inhibitor treatment
Bacteria were treated over-night in the presence of 20 µM ATR-002 or DMSO as described above. The cultures were then adjusted to OD600 = 1 in PBS, diluted 1:100 in BHI medium and incubated for 6 h at 42 °C to induce heat stress. To quantify viable bacteria the cultures were centrifuged at 4000 rpm, 4 °C for 5 min, resuspended in 1 ml PBS, serially diluted and plated on BHI agar plates.
Time-of-addition assay
For characterization of the anti-bacterial properties of the MEK-inhibitor ATR-002, an over-night culture was prepared. Sub-cultures were prepared by diluting in a ratio of 1:50 in BHI medium. Immediately after inoculation, the cultures were supplemented with DMSO, U0126 (50 µM), ATR-002 (20 µM) or gentamicin (as indicated) and the initial OD600 was measured. This measurement was repeated every three hours until 9 h post inoculation. Each culture was then diluted in BHI medium (1:500) and incubated over-night at 37 °C, 5% CO2. Finally, the OD600 of each culture was measured again.
Antimicrobial susceptibility testing
Bacterial cultures were treated over-night with 20 µM ATR-002, DMSO or medium as described above. The cultures were then centrifuged at 4000 rpm, 4 °C for 5 min and the bacterial pellet resuspended in 1 ml PBS. Antimicrobial susceptibility testing was then prepared with 100 µl aliquots of each culture on BHI agar plates, followed by application of M.I.C.Evaluator strips (purchased from Oxoid) onto the agar. Plates were incubated at 37 °C for 24 h. The concentration preventing bacterial growth was termed as the minimal inhibitory concentration (MIC) for each individual antibiotic tested.
In vitro kinase assay
For performing an in vitro kinase assay, 0.5 µg of the purified bacterial kinase domain (PknB1–291) were incubated in 30 µl kinase buffer [50 mM Tris/HCl (pH 7.5), 3 mM MgCl2, 3 mM MnCl2 and 1 mM DTT] +/− 1 µl ATP (2 mM), +/− 8 µCi 32P-ATP together with 4 µg of the substrate MBP for 1 h at 37 °C in the presence of different concentrations of ATR-002 (as indicated) or kinase buffer alone. The reaction was stopped by addition of 5 × SDS sample buffer, denaturation of the proteins at 95 °C for 10 min. Thereafter, proteins were analyzed by SDS-PAGE and Western Blot. Phosphorylated protein bands were visualized by radiography.
Statistical analysis
All experiments were performed at least three times. Results are presented as mean + SD. Statistical significance was calculated with GraphPad Prism software versions 6 using the indicated statistical tests. P-values are indicated by asterisks *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Morens, D. M., Taubenberger, J. K. & Fauci, A. S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 198, 962–70 (2008).
Iverson, A. R. et al. Influenza virus primes mice for pneumonia from Staphylococcus aureus. J. Infect. Dis. 203, 880–888 (2011).
Chertow, D. S. & Memoli, M. J. Bacterial Coinfection in Influenza A Grand Rounds Review. JAMA 309, 275–282 (2014).
McDanel, J. S. et al. Increased Mortality Rates Associated with Staphylococcus aureus and Influenza Co-Infection, Maryland and Iowa, USA. Emerg. Infect. Dis. 22, 1253–1256 (2016).
Martin-Loeches, I., Someren Gréve, F. v. & Schultz, M. J. Bacterial pneumonia as an influenza complication. Curr. Opin. Infect. Dis. 30, 201–207 (2017).
Cauley, L. S. & Vella, A. T. Why is coinfection with influenza virus and bacteria so difficult to control? Discov. Med 19, 33–40 (2015).
Li, M. et al. Evolution of virulence in epidemic community-associated methicillin-resistant Staphylococcus aureus. PNAS 20, 5883–5888 (2009).
Gould, I. M. et al. New insights into meticillin-resistant Staphylococcus aureus (MRSA) pathogenesis, treatment and resistance. Int. J. Antimicrob. Agents 39, 96–104 (2012).
Chao, D. L., Bloom, J. D., Kochin, B. F., Antia, R. & Longini, I. M. The global spread of drug-resistant influenza. J. R. Soc. Interface 9, 648–656 (2012).
Müller, K. H. et al. Emerging cellular targets for influenza antiviral agents. Trends Pharmacol. Sci. 33, 89–99 (2012).
Ludwig, S. Influenza viruses and MAP kinase cascades – Novel targets for an antiviral intervention? Signal Transduct. 7, 81–88 (2007).
Planz, O. Development of cellular signaling pathway inhibitors as new antivirals against influenza. Antiviral Res. 98, 457–468 (2013).
Hussain, M., Galvin, H., Haw, T. Y., Nutsford, A. & Husain, M. Drug resistance in influenza A virus: the epidemiology and management. Infect. Drug Resist. 10, 121–134 (2017).
Ludwig, S., Planz, O., Pleschka, S. & Wolff, T. Influenza-virus-induced signaling cascades: Targets for antiviral therapy? Trends Mol. Med. 9, 46–52 (2003).
Oviedo-Boyso, J. et al. The Phosphoinositide-3-Kinase – Akt Signaling Pathway Is Important for Staphylococcus aureus Internalization by Endothelial Cells. Infect. Immun. 79, 4569–4577 (2011).
Droebner, K., Pleschka, S., Ludwig, S. & Planz, O. Antiviral activity of the MEK-inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo. Antiviral Res. 92, 195–203 (2011).
Pleschka, S. et al. Influenza virus propagation is impaired by inhibition of the Raf / MEK / ERK signalling cascade. Nat. Cell Biol. 3, (2001).
Haasbach, E. et al. The MEK-inhibitor CI-1040 displays a broad anti-influenza virus activity in vitro and provides a prolonged treatment window compared to standard of care in vivo. Antiviral Res. 142, 178–184 (2017).
Marjuki, H. et al. Membrane Accumulation of Influenza A Virus Hemagglutinin Triggers Nuclear Export of the Viral Genome via Protein Kinase C alpha-mediated Activation of ERK Signaling. J. Biol. Chem. 281, 16707–16715 (2006).
LoRusso, P. M. et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J. Clin. Oncol. 23, 5281–5293 (2005).
Wabnitz, P. A., Mitchell, D. & Wabnitz, D. In Vitro and in Vivo Metabolism of the Anti-Cancer Agent CI-1040, a MEK Inhibitor, in Rat, Monkey, and Human. Pharmaceutical research 21, (2004).
Ratner, A. J. et al. Cystic Fibrosis Pathogens Activate Ca2+ -dependent Mitogen-activated Protein Kinase Signaling Pathways in Airway Epithelial Cells. J. Biol. Chem. 276, 19267–19275 (2001).
Seki, M. et al. Immunokinetics in severe pneumonia due to influenza virus and bacteria coinfection in mice. Eur. Respir. J. Off. J. Eur. Soc. Clin. Respir. Physiol. 24, 143–149 (2004).
Klemm, C. et al. Mitogen-activated protein kinases (MAPKs) regulate IL-6 over- production during concomitant influenza virus and Staphylococcus aureus infection. Sci. Rep. 7, 1–15 (2017).
Bakal, C. J. & Davies, J. E. No longer an exclusive club: eukaryotic signalling domains in bacteria. Trends Cell Biol. 10, 32–38 (2017).
Pereira, S. F. F., Goss, L. & Dworkin, J. Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol. Mol. Biol. Rev. 75, 192–212 (2011).
Kennelly, P. J. Protein kinases and protein phosphatases in prokaryotes: a genomic perspective. FEMS Microbiol. Lett. 206, 1–8 (2002).
Manuse, S., Fleurie, A., Zucchini, L., Lesterlin, C. & Grangeasse, C. Role of eukaryotic-like serine/threonine kinases in bacterial cell division and morphogenesis. FEMS Microbiol. Rev. 40, 41–56 (2015).
Rakette, S., Donat, S., Ohlsen, K. & Stehle, T. Structural Analysis of Staphylococcus aureus Serine / Threonine Kinase PknB. PLoS One 7, 1–9 (2012).
Burnside, K. et al. Regulation of hemolysin expression and virulence of staphylococcus aureus by a serine/threonine kinase and phosphatase. PLoS One 5, (2010).
Bugrysheva, J., Froehlich, B. J., Freiberg, J. A. & Scott, J. R. Serine/threonine protein kinase Stk is required for virulence, stress response, and penicillin tolerance in Streptococcus pyogenes. Infect. Immun. 79, 4201–4209 (2011).
Débarbouillé, M. et al. Characterization of a Serine/Threonine Kinase Involved in Virulence of Staphylococcus aureus. J. Bacteriol. 191, 4070–4081 (2009).
Donat, S. et al. Transcriptome and Functional Analysis of the Eukaryotic-Type Serine/Threonine Kinase PknB in Staphylococcus aureus. J. Bacteriol. 191, 4056–4069 (2009).
Miller, M. et al. Staphylococcal PknB as the First Prokaryotic Representative of the Proline-Directed Kinases. PLoS One 5, 1–8 (2010).
McCullers, J. a. Insights into the interaction between influenza virus and pneumococcus. Clin. Microbiol. Rev. 19, 571–582 (2006).
Niemann, S. et al. Combined action of influenza virus and Staphylococcus aureus panton-valentine leukocidin provokes severe lung epithelium damage. J. Infect. Dis. 206, 1138–48 (2012).
McCullers, J. A. The co - pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Microbiol. 1–11, https://doi.org/10.1038/nrmicro3231 (2014).
Allen, L., Sebolt-Leopold, J. & Meyer, M. B. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). Semin. Oncol. 30, 105–116 (2003).
Vornhagen, J. et al. Kinase Inhibitors that Increase the Sensitivity of Methicillin Resistant Staphylococcus aureus to β-Lactam Antibiotics. Pathogens 4, 708–721 (2015).
Tamber, S., Schwartzman, J. & Cheung, A. L. Role of PknB Kinase in Antibiotic Resistance and Virulence in Community-Acquired Methicillin-Resistant Strain USA300. Infect. Immun. 78, 3637–3646 (2010).
Kant, S., Asthana, S., Missiakas, D. & Pancholi, V. A novel STK1-targeted small- molecule as an ‘ antibiotic resistance breaker’ against multidrug-resistant Staphylococcus aureus. Sci. Rep. 7, 1–19 (2017).
Ohlsen, K. & Donat, S. The impact of serine/threonine phosphorylation in Staphylococcus aureus. Int. J. Med. Microbiol. 300, 137–141 (2010).
Lombana, T. N. et al. Allosteric activation mechanism of the Mycobacterium tubercolosis receptor Ser/Thr protein kinase, PknB. Structure 18, 1667–1677 (2011).
Smith, A. M. et al. Kinetics of coinfection with influenza A virus and Streptococcus pneumoniae. PLoS Pathog. 9, e1003238 (2013).
McCullers, J. A. & Webster, R. G. A mouse model of dual infection with inf luenza virus and Streptococcus pneumoniae. Int. Congr. Ser. 1219, 601–607 (2001).
McCullers, J. A. & Rehg, J. E. Lethal Synergism between Influenza Virus and Streptococcus pneumoniae: Characterization of a Mouse Model and the Role of Platelet-Activating Factor Receptor. J. Infect. Dis. 186, 341–350 (2002).
Strobel, M. et al. Post-invasion events after infection with Staphylococcus aureus are strongly dependent on both the host cell type and the infecting S. aureus strain. Clin. Microbiol. Infect. 22, 799–809 (2016).
Tuchscherr, L. et al. Staphylococcus aureus phenotype switching: An effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol. Med. 3, 129–141 (2011).
Fraunholz, M. & Sinha, B. Intracellular Staphylococcus aureus: live-in and let die. Front. Cell. Infect. Microbiol. 2, 43 (2012).
Bayles, K. W. et al. Intracellular Staphylococcus aureus Escapes the Endosome and Induces Apoptosis in Epithelial Cells. Infect Immun 66, 336–342 (1998).
Hardt, P. et al. The cell wall precursor lipid II acts as a molecular signal for the Ser/Thr kinase PknB of Staphylococcus aureus. Int. J. Med. Microbiol. 307, 1–10 (2017).
Liebeke, M., Meyer, H., Donat, S., Ohlsen, K. & Lalk, M. A metabolomic view of staphylococcus aureus and its ser/thr kinase and phosphatase deletion mutants: Involvement in cell wall biosynthesis. Chem. Biol. 17, 820–830 (2010).
Ludwig, S. et al. The stress inducer arsenite activates mitogen-activated protein kinases extracellular signal-regulated kinases 1 and 2 via a MAPK kinase 6/p38- dependent pathway. J. Biol. Chem. 273, 1917–1922 (1998).
Holzberg, M., Boergeling, Y., Schräder, T., Ludwig, S. & Ehrhardt, C. Vemurafenib limits Influenza A Virus propagation by targeting multiple signaling pathways. Front. Microbiol. 8, 1–13 (2017).
Hrincius, E. R. et al. CRK adaptor protein expression is required for efficient replication of avian influenza A viruses and controls JNK-mediated apoptotic responses. Cell. Microbiol. 12, 831–843 (2010).
Schoenfelder, S. M. K. et al. Methionine Biosynthesis in Staphylococcus aureus Is Tightly Controlled by a Hierarchical Network Involving an Initiator tRNA-Specific T-box Riboswitch. PLoS Pathog. 9, 1–9 (2013).
Acknowledgements
We would like to thank Prof. Dr. Sven Hammerschmidt for kindly providing S. pneumoniae strains used in the present study. Furthermore, we thank Danielle Brandes and Chairini Cassia Thome for great technical assistance during their practical courses and ATRIVA Therapeutics GmbH to provide CI-1040 and ATR-002. This work was supported by the Deutsche Forschungsgemeinschaft (grants SFB 1009 projects B01 and B02, DFG Lu477/25-1, SFB TR34 project A2), the Innovative Medical Research fund EH121307, the IZKF (grants EhC2/006/15 and Lud2/008/17), the Cells in Motion Excellence Cluster (CiM) and the MedK Muenster. We gratefully acknowledge the support by the Open Access Publication Fund of the University of Muenster.
Author information
Authors and Affiliations
Contributions
C.B. and C.E. conceived and designed the experiments. C.B., M.J., C.M. and T.H. performed the experiments. C.B., C.E. and S.L. analysed the data and wrote the manuscript. S.N., G.P., O.P. and K.O. provided tools. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bruchhagen, C., Jarick, M., Mewis, C. et al. Metabolic conversion of CI-1040 turns a cellular MEK-inhibitor into an antibacterial compound. Sci Rep 8, 9114 (2018). https://doi.org/10.1038/s41598-018-27445-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27445-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
