
Abstract
The loss of endogenous cardiac regenerative capacity within the first week of postnatal life has intensified clinical trials to induce cardiac regeneration in the adult mammalian heart using different progenitor cell types. We hypothesized that donor age-related phenotypic and functional characteristics of cardiac progenitor cells (CPC) account for mixed results of cell-based cardiac repair. We compared expression profiles and cell turnover rates of human heart-derived c-kitpos progenitors (c-kitpos CPC) and cardiosphere-derived cells (CDC) from young and adult donor origin and studied their in vitro angiogenic and cardiac differentiation potential, which can be relevant for cardiac repair. We report that 3-dimensional CDC expansion recapitulates a conducive environment for growth factor and cytokine release from adult donor cells (aCDC) that optimally supports vascular tube formation and vessel sprouting. Transdifferentiation capacity of c-kitpos CPCs and CDCs towards cardiomyocyte-like cells was modest, however, most notable in young c-kitpos cells and adult CDCs. Progenitors isolated with different methods thus show cell- and donor-specific characteristics that may account for variable contributions in functional myocardial recovery.
Similar content being viewed by others

Introduction
The adult mammalian heart is traditionally considered as a terminally differentiated organ, not able to regenerate after massive cell loss. Acute myocardial infarction (AMI), the most severe presentation of ischemic heart disease, remains the leading cause of death worldwide (2013, WHO) and generally implies the loss of approximately 1 billion cardiac myocytes (CM). In survivors of large AMI involving more than 25–30% of left ventricular mass, the heart undergoes a remodelling process with progressive dilatation and functional impairment, resulting in heart failure (HF). The HF syndrome1 affects 4% of the population worldwide and carries an ominous prognosis despite state-of-the-art guideline-recommended therapies2, emphasizing the need for innovative treatments. To date, the safety and efficacy of multiple candidate cell types, including skeletal myoblasts3, heterogeneous bone marrow-derived mononuclear cells4 and mesenchymal stem cells (MSC)5 have been studied in preclinical and clinical trials of ischemic myocardial damage with mixed results and overall low rates of progenitor cell engraftment and cardiac differentiation. The more recent discovery of endogenous cardiac progenitor cells (CPC) together with the observation of a persistent, yet limited regenerative potential in the adult heart6, prompted a shift towards CPCs as a promising candidate for cell-based therapeutic interventions.
In contrast to most tissues, the heart is host to a remarkably extended list of progenitor cells, a discovery that is based on a multiparametric strategy for their phenotypic and functional characterization. The use of different cell surface receptors (c-kit7, Sca18), lineage marker cocktails, dye expulsion characteristics typical of side population (SP) phenotype with long-term repopulation capacity (SP cells)9 and (non-) adherent growth properties in culture (MSCs5, cardiospheres and cardiosphere-derived cells (CDC)10 have revealed variable levels of cardiac commitment, not paralleling the capacity for cardiac repair. To what extent this is caused by different transcriptional profiles or by donor age-related functional impairment of heart-derived progenitor cells, remains incompletely understood. The molecular signature of some of these culture expanded heart-derived progenitors has recently been compared in age- and gender-matched mice11, but has not yet been established in humans. In this study, we therefore focused on 2 heart-derived progenitor cell populations that have been recently introduced in clinical translation, autologous c-kitpos–selected CPCs12 and CDCs13,14. For the first time we compared molecular signatures and proliferation and differentiation characteristics of progenitor cells from young donor hearts (c-kitpos yCPC and yCDC) with cells obtained from adult donors with advanced ischemic disease (c-kitpos aCPC and aCDC). Here we report that culture expanded CDCs derived from adult donors have a distinct transcriptional profile with greater cell cycle activity and prominent in vitro paracrine growth factor release. The molecular signature of aduIt CDCs favours pro-angiogenic, cytotrophic and immunomodulatory effects while cardiac transdifferentiation potential is modest and most notable in c-kitpos yCPCs and aCDCs.
Results
Morphological characterization of human adult and young c-kitpos CPCs and CDCs
To derive progenitor cells from human right atrial (RA) appendage biopsies, two different isolation methods were used (Fig. 1a): positive selection using the surface marker c-kit (CD117) to isolate c-kitpos CPCs and formation of cardiospheres after two weeks of explant culture to derive 2D cultures of CDCs. Phenotypically, CDCs represent a more heterogeneous population of cells irrespective of donor age (young versus adult) compared to c-kitpos cells, but no major differences in shape or size were observed between progenitor cells from young and adult donor hearts at early passages (Fig. 1b) as confirmed with FACS analysis based on FSC plots (data not shown).

Isolation and morphological comparison of c-kitpos CPCs and CDCs. (a) Schematic overview of the steps involved to isolate c-kitpos CPCs and CDCs. To isolate c-kitpos cells, a human RA appendage biopsy is minced into small pieces and collagenase digested to culture a heterogeneous mixture of single cells for 2 passages prior to MACS-based selection. To obtain CDCs, a mild collagenase-based digestion is performed on small myocardial fragments to allow outgrowth of fibroblasts and phase-bright cells from the cardiac explants. The latter are transferred onto a poly-D-Lysine coated dish to form cardiospheres. Finally, the cardiospheres are collected and grown as monolayer cells called CDCs. (b) Phase bright-field pictures of c-kitpos CPCs and CDCs derived from young and adult donors. Scale bars represent 200 µm. CPC, cardiac progenitor cell; CDC, cardiosphere-derived cell; RA, right atrial.
Gene expression profiling of CDCs and c-kitpos CPCs derived from adult and young donors
To identify biological differences between different progenitor cell populations, we performed RNA sequencing and pathway analysis of CDCs and c-kitpos CPCs derived from adult and young donors. The full list of differentially expressed transcripts and identified processes and pathways is available as an Online Supplement. Based on hierarchical clustering, the greatest contrast was observed between aCDCs and yCDCs and between aCDCs and c-kitpos CPCs derived from young and adult donors (Fig. 2a). In total, 2,886 transcripts showed a significant expression difference of at least 2-fold between aCDCs and yCDCs: 1,423 had higher expression levels and 1,463 had lower expression levels. String network analysis revealed clusters of transcripts related to cytokines and inflammation, growth factors, cell motility and extracellular matrix (Fig. 2b). A second independent approach using gene ontology analysis confirms significant enrichment of biological processes related to the inflammatory response, cell growth, angiogenesis and cell migration in aCDCs compared to c-kitpos aCPCs (Fig. 2c, blue) and enrichment for cell cycle processes, angiogenesis and cell proliferation when comparing aCDCs to yCDCs (Fig. 2d, blue). We then performed pathway analysis and quantified the activation of the identified canonical pathways. More specifically, this revealed significant activation of Toll-like Receptor, TREM1 and IL-8 signalling in aCDCs compared to c-kitpos aCPCs (Fig. 2c, green), and cell cycle related pathways in aCDCs compared to yCDCs (Fig. 2d, green). In addition, eNOS and glioma invasiveness signalling were more activated whereas LXR/RXR and PPARα/RXRα activation, GnRH (Gonadotropin Releasing Hormone) signalling, α-adrenergic, and cardiac hypertrophy signalling were significantly deactivated in aCDCs (Fig. 2c and Table 1).

Differential gene expression between CDCs and c-kitpos CPCs derived from adult and young donors. (a) Heat map of Spearman correlations with the greatest differences observed between aCDCs and c-kitpos CPCs and between aCDCs and yCDCs. (b) String network analysis using the top significant genes showed clusters of transcripts that are related to cytokines and inflammation, growth factors, cell motility and extracellular matrix. (c,d) Gene ontology results using DAVID software (blue) and pathway analysis (IPA) (green) comparing aCDCs and c-kitpos aCPCs (c) and yCDCs and aCDCs (d) confirming the string network analysis. Compared to c-kitpos aCPCs, aCDCs are more enriched in inflammatory processes (c), while cell cycle pathways are more prominent in aCDCs compared to yCDCs (d). (e–g) Heatmaps of immunomodulatory genes, cell cycle markers and stem cell markers and growth factors, red = downregulated; blue = upregulated. c-kitpos yCPC (ages 5 d, 2 m, 2.5 m, 3 m, 4 m), c-kitpos aCPC (ages 62 y, 65 y, 74 y, 79 y, 82 y), yCDC (ages 4 d, 1,5 m, 3 m, 5 m, 10 y), aCDC (ages 47 y, 65 y, 70 y, 75 y, 77 y).
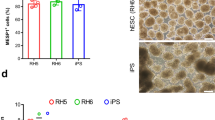
Subsequently, we investigated the identified functional differences on a gene expression level for immunomodulation (Fig. 2e), cell cycle markers (Fig. 2f) and stem cell markers and growth factors (Fig. 2g). With respect to immunomodulation, many inflammatory genes including IL1B, IL8 (CXCL8) and TGFB1 were higher expressed in aCDCs compared to yCDCs, but also when compared to c-kitpos aCPCs (Fig. 2e). The difference between CDCs and c-kit-selected CPCs is predominantly driven by greater inflammatory signalling (Toll-like receptors TLR, TREM1, IL8) with the highest activation in aCDCs, an intermediate profile in yCDCs, and the lowest expression levels in c-kitpos CPCs irrelevant of donor age. Other transcripts including IL18, TGFB2 and TLR4, however, are lower expressed in aCDCs compared to yCDCs. The expression profile of cell cycle-related transcripts in aCDCs is consistent with greater proliferation potential when compared to yCDCs, while this difference appears to be less pronounced in c-kit-selected CPCs with a greater proliferation potential for the c-kitpos yCPCs (Fig. 2f). For the transcription factors, only MEF2C was higher expressed in aCDCs compared to yCDCs and c-kitpos CPCs isolated from adult and young donors. In contrast, the expression of MEST, a member of the hydrolase superfamily important in early mesoderm formation, is the lowest in aCDCs while TBX5 and GATA4 had comparable transcript levels in all progenitor cell populations. The expression of angiogenic growth factors CXCL12 (stromal-derived factor-1 alpha (SDF1-α)), HGF (hepatocyte growth factor), PlGF (placental growth factor), VEGFA (vascular endothelial growth factor A) and FGF2 (fibroblast growth factor 2, or basic fibroblast growth factor) is more highly expressed in aCDCs and to a lesser extent in yCDCs compared to c-kitpos aCPCs and c-kitpos yCPCs. Finally, the stem cell factors KIT and KLF4 are the lowest in aCDCs compared to yCDCs and c-kitpos CPCs derived from young and adult donors (Fig. 2g).
Taken together and when compared to c-kitpos aCPCs, aCDCs display a higher activation of cell cycle pathways, inflammatory pathways important for reparative responses (TLR, TREM1, Pattern Recognition Receptors), critical growth factors and eNOS signalling, all of which may contribute to a unique cardioprotective capacity during cytotoxic stress conditions.
Gene signature and proliferation capacity of heart-derived c-kitpos CPCs and CDCs
To confirm RNA sequencing expression data, we performed qPCR analysis and measured significantly higher CD117 (c-kit) transcript levels in c-kitpos yCPCs and c-kitpos aCPCs compared to aCDCs (Fig. 3a) and within the CDC population no significant difference was found in CD117 levels. The fibroblast marker DDR2, was equally expressed in young and adult c-kitpos CPCs and CDCs. The expression of the proliferation-associated and hypoxia-inducible endoglin CD105 was prominent in all four cell populations but significantly higher in aCDCs compared to yCDCs and c-kitpos aCPCs (p < 0.05). Conversely, aCDCs showed significantly lower gene expression level of the mesenchymal marker CD90 compared to yCDCs and c-kitpos yCPCs and c-kitpos aCPCs (p < 0.05). To study lineage commitment of human progenitor cells, expression levels of the endothelial cell (EC) marker CD31, the early cardiac transcription factors MEF2c and GATA4 and the smooth muscle cell (SMC) marker MYH11, were measured. Interestingly, CD31 and MEF2c expression was significantly higher in aCDCs compared to yCDCs and c-kitpos CPCs derived from young and adult donors (p < 0.05). The same trend was observed for GATA4 expression however the expression was only significant compared to c-kitpos yCPC. The expression level of the SMC marker MYH11 was significantly higher in cells derived from young donors compared to cells derived from adult donors irrespective of the isolation method (p < 0.05).

Phenotypic comparison of adult and young c-kitpos CPCs and CDCs. (a) Gene expression levels of CD117 (KIT), CD105, CD90, the EC marker CD31, early cardiac transcription factors GATA4 and MEF2c, SMC marker MYH11 and fibroblast marker DDR2. c-kitpos yCPC (n = 8), c-kitpos aCPC (n = 9), yCDC (n = 7) and aCDC (n = 7). Results are geometric mean with 95% CI, one-way ANOVA was performed for normally distributed data and Kruskal-Wallis test for non-normally distributed data, *p < 0.05. EC, endothelial cell; SMC, smooth muscle cell; CPC, cardiac progenitor cell; CDC, cardiosphere-derived cell. (b) Flow cytometry analysis of c-kitpos yCPCs (n = 8), c-kitpos aCPCs (n = 10), yCDCs (n = 7) and aCDCs (n = 8) for CD90, CD105, and CD117. All analyses were performed after 6 passages. Results are mean ± SEM, *p < 0.05. (c) PDT of c-kitpos yCPCs (n = 4), c-kitpos aCPCs (n = 5), yCDCs (n = 5) and aCDCs (n = 4). Results are mean ± SEM, *p < 0.05. CPC, cardiac progenitor cell; CDC, cardiosphere-derived cell; PDT, population doubling time.
Because heart-derived progenitor cells have important paracrine effector mechanisms, we compared gene expression of different candidate growth factors and cytokines. Transcript levels of FGF2 were significantly higher in yCDCs and aCDCs than in c-kitpos yCPCs (p < 0.05). In addition, the expression of HGF and PlGF, was significantly higher in aCDCs compared to c-kitpos yCPCs and c-kitpos aCPCs (p < 0.05 and p < 0.01, respectively). Finally, IGF-1 and insulin growth factor-receptor2 (IGF-2R) gene expression were significantly higher in aCDCs than in c-kitpos aCPCs (p < 0.05; Fig. S1). Also, the expression of inflammation markers IL1B, IL8 and TGFB1 were higher in aCDCs compared to yCDCs, which confirms the RNA sequencing data (Fig. S2). In addition, TGFB1 expression was highest in aCDCs compared to yCDCs, c-kitpos yCPCs and c-kitpos aCPCs (data not shown).
The presence of the selection marker c-kit after 5 to 6 passages in culture was also verified using flow cytometry analysis (Fig. 3b). Consistent with quantitative gene expression data, c-kitpos CPCs from adult and young donors expressed CD117 (KIT) to variable degrees (53 ± 7% and 35 ± 8%, respectively). In contrast, CD117 was almost undetectable in aCDCs (0.2 ± 0.1%) and very low in yCDCs (3.4 ± 2.0%). Cell surface expression of the mesenchymal marker CD90 (THY-1) was significantly higher in c-kitpos CPCs (aCPC, 91 ± 2%; yCPC, 84 ± 3%) than in aCDCs (22 ± 3%, p < 0.05) and in addition significantly higher in c-kitpos aCPC compared to yCDCs (58 ± 10%). This protein plays an important role in cell-cell and cell-matrix interactions and expression levels have been reported to be inversely related to regenerative capacity of CPCs15. CD105 protein, an accessory TGF-beta receptor involved in angiogenic signalling, was expressed on the surface of virtually all progenitors but to a lesser extent on CDCs (c-kitpos yCPC, 99 ± 0.3%; c-kitpos aCPC, 99 ± 0.2%; yCDC, 67 ± 3.0%; aCDC, 88 ± 4%; yCDC compared to c-kitpos CPCs p < 0.001; aCDC versus c-kitpos yCPC p < 0.05).
To evaluate proliferation capacity of the different progenitor cell types, we calculated population-doubling times (PDT) (Fig. 3c). Consistent with observed differences in transcript levels of cell-cycle regulated genes (Fig. 2f), c-kitpos yCPCs have a significantly lower PDT and thus higher proliferation rate compared to c-kitpos aCPCs (2.25 ± 0.10 days versus 3.62 ± 0.24 days, p < 0.02). In contrast, adult progenitor cells isolated according to the explant culture technique (aCDC) showed significantly lower PDTs and thus higher proliferation rates than yCDCs (1.78 ± 0.11 days versus 6.37 ± 0.66 days, p < 0.02). In addition, the average passage number at which proliferation of the cells stagnated or started to decline, was significantly lower in c-kitpos aCPCs and yCDCs (13.0 ± 0.7 and 12.1 ± 0.7 passages, respectively), the cell groups with the highest PDTs, compared to c-kitpos yCPCs and aCDCs (21.3 ± 3.0 and 22.0 ± 0.4 passages, respectively).
Vasculogenic and angiogenic potential of c-kitpos CPCs and CDCs derived from adult and young donors
Next, the vasculogenic potential of human progenitors was evaluated after 6 hours culture of the cells on Matrigel® (Fig. 4a–c). To develop firmly established vascular networks, progenitor cells must be able to sprout, form branches and interconnect them into segments, bounded by a junction or node (Fig. 4a). The number of branches was similar between the 4 different cell populations (c-kitpos yCPC, 6.1 ± 1.4/mm2; c-kitpos aCPC, 7.6 ± 0.8/mm2; yCDC, 5.76 ± 0.17/mm2; aCDC, 5.2 ± 0.5/mm2). However, the ability to form vascular networks, as reflected by the number of nodes, was significantly lower for c-kitpos yCPCs (c-kitpos yCPC, 23.0 ± 5.6/mm2) compared to progenitor cells isolated from adult donors (aCDC, 57.1 ± 8.1/mm2). Of note, yCDCs show a trend for a higher tube formation potential when compared to c-kitpos yCPCs (Fig. 4a,b). These relative differences in vascular network formation may in part be attributable to variable CD90 mRNA and protein expression patterns (Fig. 3), which have been shown in previous studies to inversely correlate with endothelial transdifferentiation and regeneration capacity15. The CD90 expression in yCDCs varied between the different donors (Fig. 3). To investigate if variation in CD90 expression is associated with differences in tube formation capacity, we measured the correlation between CD90 mRNA expression and the number of nodes. Here, we report an inverse correlation between the number of nodes and CD90 mRNA expression of aCDCs and yCDCs (Fig. 4c). Because the in vitro Matrigel® assay does not allow evaluation of vascular sprouting and formation of lumen structures, a fundamental property of ECs, we complemented our analysis with an in vitro 3D spheroid assay in Matrigel® to study sprouting angiogenesis (Fig. 4d). The additional 3D spheroid assay confirmed the superior potential of aCDCs (n = 2) to sprout and form a vascular network compared to yCDCs (n = 2), c-kitpos yCPCs (n = 2) and c-kitpos aCPCs (n = 2). Taken together, our data indicate that all human progenitor types have an intrinsic vasculogenic potential, however, progenitors isolated from young donors are possibly more immature, with reduced endothelial lineage commitment resulting in less developed vascular networks.

Vasculogenic and angiogenic potential of adult and young c-kitpos CPCs and CDCs. (a) Analysis of network formation 6 hours after culture on Matrigel® of c-kitpos yCPCs (n = 5), c-kitpos aCPCs (n = 8), yCDCs (n = 5) and aCDCs (n = 9). (b) Quantification of number of nodes, junctions, meshes, segments and branches/mm2. Results are mean ± SEM, *p < 0.05. (c) Correlation of vasculogenic potential, expressed as number of nodes/mm2 and CD90 mRNA expression of aCDCs and yCDCs. (d) Representative images of 3D sprouting of c-kitpos yCPC (n = 2), c-kitpos aCPC (n = 2), yCDC (n = 2) and aCDC (n = 2) spheroids after 48 hours incubation. Per group, cells of 2 different donors were used and for each donor, 4 different spheroids were imaged. One representative spheroid per donor is depicted. (e) Growth factor release analysis in conditioned medium collected after 24 hours incubation of c-kitpos yCPCs (n = 9), c-kitpos aCPCs (n = 11), yCDCs (n = 7) and aCDCs (n = 10) for IGF-1, VEGF, bFGF, PlGF, HGF and SDF-1. Results are mean ± SEM, *p < 0.05 compared to aCDC and #p < 0.05 compared to yCDC. CPC, cardiac progenitor cell; CDC, cardiosphere-derived cell.
To further explore the angiogenic phenotype, growth factor secretome was analysed in medium conditioned by human CPCs using ELISA for IGF-1, bFGF, HGF, PlGF, VEGF and SDF-1 (Fig. 4e). Consistent with qRT-PCR data, the secretion of IGF-1, HGF, bFGF, VEGF and PlGF was significantly higher in CDCs compared to c-kitpos CPCs, irrespective of donor age (*p < 0.05 versus aCDC; #p < 0.05 versus yCDC). In addition, within the CDC population the secretion of IGF-1 was significantly lower in cells derived from young hearts. Of note, the secretion of the chemotactic protein SDF-1, a key regulator of stem cell migration to sites of tissue injury16, was undetectable in c-kitpos CPCs derived from adult and young donors and in yCDCs but prominent in the secretome obtained from aCDCs (p < 0.01).
Finally, because of the recently recognized important role of exosomes, small extracellular membrane vesicles containing proteins, mRNA and miRNA that are secreted by cells to induce cellular communication, we measured exosome levels in 48 hours serum-free conditioned medium. Nanoparticle analysis technology to determine the concentration and particle distribution demonstrated that exosomes derived from aCDCs and yCDCs represented a more homogeneous and purified population than those shed by c-kitpos aCPCs and yCPCs and contain variable concentrations of defined miRNAs with a presumed cyto- or cardioprotective effect17 (Fig. S3A,B).
Cardiac differentiation potential of c-kitpos CPCs and CDCs derived from adult and young donors
To test the hypothesis that heart-derived progenitors from young donors have a greater cardiac differentiation potential than progenitors derived from adult donors, all cell populations were incubated with cardiac differentiation medium (DIFF) or regular expansion medium (CON). Immunofluorescence stainings showed the presence of the cardiac contractile protein α-sarcomeric actinin in CDCs and c-kitpos CPCs after 12 days incubation with DIFF medium (Fig. 5a,b). Contrary to our hypothesis, we observed a greater increase of α-sarcomeric actinin-positive cells (DIFF compared to CON) under these culture conditions in aCDCs compared to yCDCs. However, the opposite effect was observed in ckitpos CPCs, with a greater increase of α-sarcomeric actinin-positive cells in c-kitpos yCPCs compared to c-kitpos aCPCs (Fig. 5b). This suggests a greater cardiac differentiation potential of c-kitpos yCPCs compared to c-kitpos aCPCs, yCDCs and aCDCs. Of note, no contractile phenotype was observed after 12 days incubation with DIFF medium. In addition, expression of the cardiac lineage markers GATA4, MEF2c and NKX2.5 confirmed activation of the cardiac differentiation program in all cells treated with cardiac differentiation medium (DIFF) (Fig. 5c), which is consistent with the increased α-sarcomeric actinin-positive cells following incubation in DIFF medium (Fig. 5a,b).

The cardiac differentiation capacity of adult and young c-kitpos CPCs and CDCs. (a) Double immunofluorescence staining of phalloidin (green) and α-sarcomeric actinin (red) revealed protein expression of a cardiac contractile protein after incubation in cardiac differentiation medium (DIFF) or regular expansion medium (CON) for 12 days. c-kitpos yCPCs (n = 5), c-kitpos aCPCs (n = 6), yCDCs (n = 5) and aCDCs (n = 5). Scale bars represent 100 µm. (b) Quantification of the immunofluorescent staining for α-sarcomeric actinin positive cells after 12 days incubation in DIFF medium. Fold change of alpha-sarcomeric actinin positive cells in DIFF treated groups compared to CON groups, results are mean ± SEM, *p < 0.05. (c) Fold change expression of GATA4, MEF2c and NKX2.5 in DIFF treated groups compared to CON groups. Results are mean ± SEM.
Culture expansion conditions do not explain the observed differences in cell phenotype and functional activity between CDCs and c-kitpos CPCs
Finally, the cell phenotype and functional activity was investigated after culturing CDCs (yCDC, n = 2; aCDC, n = 2) in c-kitpos CPC expansion medium and c-kitpos CPCs (c-kitpos yCPC, n = 2; c-kitpos aCPC, n = 2) in CDC expansion medium on fibronectin-coated culture plates for at least 2 passages. The expression levels of CD117, CD105 and CD90 in these cells cultured in their counterpart medium (SWAP) was tested with qPCR and compared to the original transcriptome as reported in Fig. 3a (Fig. S4A). We measured similar expression profiles for all 3 genes irrespective of the expansion medium with the highest expression of CD117 in c-kitpos yCPCs. CD105 was equally expressed in all cell groups and CD90 showed again the lowest expression in aCDCs. Next, the vasculogenic potential of the four cell groups was explored after culturing the progenitors with their counterpart medium (SWAP). Again, results showed that the ability to form vascular networks, as reflected by the number of nodes, was lower in progenitors isolated from young donors compared to adult donors (Fig. S4B). In addition and again in concert with our original observations (Fig. 4a,b), the number of junctions, meshes, segments and branches showed a comparable pattern with progenitors derived from adult donors forming more complex networks compared to progenitors derived from young donors. In conclusion, these data suggest that CDC and c-kitpos CPC culture conditions do not account for the phenotypic and functional differences between the four progenitor groups.
Discussion
Recent evidence indicates that the mammalian heart harbours multiple cardiogenic progenitor cell populations and stages a marked endogenous regenerative response after tissue damage in early postnatal life18. Because this capacity for scar-free healing through cardiomyogenesis is lost in the adult heart, intracardiac transfer of c-kitpos CPCs and a heterogeneous explant population of CDCs has been introduced in clinical trials in an effort to enhance cardiac repair after ischemic injury. In the present study, we aimed to bridge the gap in our understanding of the molecular signature of these 2 human heart-derived progenitor cell populations obtained at different stages in postnatal life. We compared in vitro the phenotypic, functional and genetic characteristics of heart-derived progenitor cells from neonates and young patients with congenital heart disease undergoing corrective surgery and from adult patients with advanced ischemic and valvular disease. We deliberately studied culture expanded cells after 4 to 5 passages as was peformed in pilot clinical studies (e.g. SCIPIO12 and CADUCEUS13,14 trials) and observed a distinct molecular signature between CDCs of different donor age, but not between c-kit-selected progenitor cells of young and adult patients. The molecular pathways with different states of activation or repression between adult and young CDCs play a pivotal role in cell survival, proliferation and endothelial cell (EC) or cardiac myocyte-specific functions. Importantly, adult heart-derived and culture expanded CDCs showed significantly higher activation of the EC-associated NOS3 gene, showed less CD90 surface gene expression and secreted high levels of pro-angiogenic cytokines and growth factors. These phenotypic characteristics conferred greater capacity for vascular tube formation and sprouting in a 3D spheroid assay. Transcriptional profiling also markedly contrasted aCDCs with c-kitpos CPCs from young and adult heart as evidenced by the consistent activation of cell cycle regulatory genes and of interest, a robust induction of immunomodulatory genes.
Transcriptome analysis of aCDCs revealed a baseline upregulation of myocyte-specific transcription factors (GATA4 and MEF2c) and the EC-specific marker (CD31), a constituent of the endothelial intercellular junction, also designated as PECAM-1 (platelet endothelial cell adhesion molecule-1). Adult CDCs also expressed significantly higher levels of endoglin or CD105, known to play a crucial role in angiogenesis19, but contrary to MSCs20 co-expression of CD90 was significantly lower. CD90 expression is lower in aCDCs compared to yCDCs, c-kitpos yCPCs and c-kitpos aCPCs. In fact, it has previously been shown that injection of CDCs expressing high levels of CD90, is associated with a reduced reparative capacity and in contrast CD90neg CDCs induce a more pronounced therapeutic effect15. Of interest, this inverse association is also clearly evidenced in our in vitro network formation assay where aCDCs induce markedly complex vascular network development. Taken together, transcriptional analysis suggests that heterogeneous aCDCs express early lineage-specific genes, some of which recapitulate an in vivo pattern in newly formed cells derived from activation and commitment of resident progenitor cells after injury21.
Somewhat unexpectedly, aCDCs had also a significantly lower PDT and thus faster cell turnover compared to their counterparts from young hearts, but this observation was consistent with the significantly higher expression of cyclins and cell cycle regulatory genes. The faster cell turnover in aCDCs than in yCDCs was not recapitulated in c-kitpos CPCs, where cells from adult donors showed a significantly slower cell turnover than c-kitpos yCPCs. A recent study showed that human Isl1pos c-kitpos CPC isolated from adult patients harbor an altered miRNA expression profile that correlated with a reduced proliferation capacity and abrogated response to growth factors compared to Isl1pos c-kitpos CPC derived from neonates22. Another study, comparing CDCs derived from patients of various ages, showed limited differences in proliferation capacity, angiogenic potential and expression of growth factors between CDCs from young and elderly patients (≥65 years)23. However, in this study the youngest donor was 2 years old, the median age 72.5 years and 61.5% of them were 65 years or older which is in striking contrast with our study. The CDCs derived from young patients were probably underrepresented compared to the CDCs derived from elderly patients to see significant differences whereas in our study CDCs from much younger donors were studied. In contrast, S. Kaushal and co-workers compared CDCs and c-kitpos CPCs derived from donors with various ages, and reported a greater in vivo regenerative capacity and a higher secretion of angiogenic factors in CDCs derived from neonates (<30 days old)24,25. In our study, we only have 1 and 2 neonates included in yCDC and c-kitpos yCPC, respectively, and this underrepresentation might account for the different results. Also, CDCs from different young ages (neonates, infants and children) showed a comparable in vivo regenerative potential26. However, in contrast to our study, the angiogenic potential was not investigated in this study. Several studies showed that cell-based therapy improves cardiac function through paracrine mechanisms (mediation of angiogenesis, anti-apoptosis and recruitment of endogenous progenitors) since incorporation of injected cells is rare event10,27. In this study, aCDCs show a broad expression panel of angiogenic cytokines and potent cytoprotective growth factors, including IGF-1, HGF, bFGF, PlGF, VEGF, and SDF-1, all of which may mediate the paracrine effects of CDCs. Also, heart-derived CDCs displayed a strikingly distinct exosome release pattern when compared to CPCs. These exosomes gained a lot of interest in view of their important role in paracrine and autocrine signalling28. In addition, injection of cardiac progenitor-derived exosomes in an AMI rat model protected CM against apoptosis, showed enhanced angiogenesis and improved cardiac function29. However, follow-up studies need to examine to what extent exosome-mediated signalling can affect molecular and cellular pathways that are critically important for cardiac repair or protection. Also, a direct correlation of a particular secreted factor(s), the importance of exosomes as a mediator and in vitro tube formation was not performed in this current study.
Finally, we investigated the cardiogenic potential of CDCs and c-kitpos CPCs in an in vitro differentiation assay. C-kitpos CPCs derived from young donors and aCDCs showed a tendency for greater transdifferentiation capacity into cardiomyocyte-like cells than c-kitpos aCPCs and yCDCs. The greater commitment towards cardiac lineages for c-kitpos yCPCs and aCDCs and the marked immunomodulatory signature and neovascularization potential of aCDCs represent interesting characteristics for future cell transfer strategies. In chronic ischemic cardiomyopathy, the maladaptive immune activation that accompanies ischemic dysfunction would require a combined intervention that at the same time mitigates excessive immune activation while supporting neovascularization and cardiomyogenesis. Recent reports have called our attention for immunomodulatory properties of cardiac progenitors that are capable of generating an antioxidant, anti-fibrotic, and anti-inflammatory environment30. Additionally, de Couto et al. showed that injection of CDCs or CDC exosomes in a rat AMI model reduces the number of pro-inflammatory macrophages and simultaneously polarizes the macrophages into a cardioprotective phenotype resulting in less CM apoptosis and reduced infarct size31,32. We observed greater activation of some inflammatory transcripts in aCDCs compared to yCDCs and c-kitpos CPCs, while other transcripts including TLR4, implicated in post-infarction inflammation33, were less expressed. Pro-inflammatory cytokines IL1B and IL8 are both more highly expressed in aCDCs and promote inflammatory leukocyte recruitment and activation, while IL1 delays myofibroblast activation and the potent pro-angiogenic IL8 contributes to infarct neovascularization34. TGF-β is a key player in the transdifferentiation of fibroblasts into myofibroblasts which secrete ECM proteins to form fibrotic scar tissue35. We measured higher TGFB1 expression in aCDCs compared to yCDCs and c-kitpos CPCs, but lower TGFB2 levels. IL18 stimulates IFN-γ secretion by NK-cells and T-cells which leads to stimulation of pro-inflammatory macrophages36. Our data show that IL18 transcripts are lower in aCDCs compared to yCDCs and c-kitpos CPCs. Taken together, the biological significance of these variable inflammatory gene expression patterns remains incompletely understood and requires targeted in vivo studies in the context of cardiac dysfunction.
Several limitations need to be considered in our study. First, the limited amount of RNA available from human progenitor cells precludes transcriptional analysis of a wider panel of genes using qRT-PCR. Second, size constraints of myocardial biopsies precluded cell biology studies on freshly isolated and non-passaged cells and prompted standardized analysis on multi-passaged cells with similar expansion profiles and passage numbers as used in clinical trials. At the same time, limited tissue availability did not allow parallel isolation protocols for both cell types from identical tissue samples. Although ex vivo culture expansion may affect genetic signatures, the consistency of the data reported in our analysis irrespective of expansion medium likely reflects the imprinted phenotype of the cells as used in recent clinical trials. Third, the great age range present in the c-kitpos yCPC and yCDC groups and the underrepresentation of neonates may mask the potential of c-kitpos CPCs and CDCs derived from neonates, which should be addressed in further studies. Fourth, we acknowledge the importance of unraveling the factors responsible for the functional effects in our in vitro studies, which needs to be addressed in follow-up studies. Finally, additional mechanisms operate in vivo including stimulation of resident cardiac cells and cytoprotective effects on ischemic cells, which have not been tested directly in this in vitro study.
In conclusion, phenotypic analysis of aCDCs reveals paracrine and immunomodulatory effects that appear to be distinct from those in yCDCs. In addition, c-kitpos yCPCs and aCDCs comprise a greater differentiation potential into cardiomyocyte-like cells. Whether these in vitro cell-specific findings correlate with clinically significant changes and whether combination of different progenitor cell types may confer greater clinical benefit than either cell type alone, needs to be addressed in targeted studies.
Methods
Tissue samples
The tissue procurement protocol was approved by the Medical Ethics Committee of the University Hospitals Leuven (study number S56078) and all experiments were performed in accordance with the Medical Ethics Committee guidelines. Additionally, informed consent was provided by all adult patients (47–84 years old; c-kitpos aCPC, n = 12; aCDC, n = 9) and by legal guardians or parents for all young patients (1 day – 14 years old; c-kitpos yCPC, n = 10; yCDC, n = 7). Main patient characteristics are shown in Tables 2 and S3.
Isolation and culture of human progenitor cells
Cardiac tissue samples were obtained from the RA appendage that was routinely discarded from patients undergoing intra-cardiac surgeries. Patients undergoing extra-cardiac surgery were excluded. All adult patients were referred for valve surgery, assist device implantation or heart transplantation. Neonatal RA appendage specimens were collected during corrective surgery for congenital heart disease. No other specific inclusion criteria were used. Human c-kitpos (CD117) CPCs were obtained after collagenase-based digestion of a RA appendage tissue sample based on the methods used in the SCIPIO trial12. Briefly, adipose tissue was removed and tissue samples were minced into small pieces before collagenase-based (Collagenase NB4 Standard Grade from Clostridium histolyticum, Serva Electrophoresis) digestion in Ham’s F12 medium containing 1–3 mg/mL collagenase. The supernatant containing single cells were collected and plated in Ham’s F12 medium (Lonza) supplemented with 10% FBS (ThermoFisher Scientific), 1% penicillin/streptomycin (5000 units/mL penicillin, 5000 µg/mL streptomycin; ThermoFisher Scientific), 5 mU/mL human erythropoietin (EPO) (Sigma-Aldrich). When 80–90% confluency was reached, positive selection for c-kit by magnetic-activated cell sorting (MACS) (CD117 microbead kit, Miltenyi Biotec) was performed. Medium was changed twice a week and cells were passaged when 80–90% confluency was reached. Human CDCs were derived according to the protocol reported by Marbán et al.10 with modifications. RA appendage tissue samples were minced and mildly digested with collagenase (Collagenase NB4 Standard Grade from Clostridium histolyticum, Serva Electrophoresis). Afterwards explants were generated and plated on fibronectin-coated (25 µg/mL) culture plates in cardiac explant medium (CEM) (Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 20% FBS, 1% penicillin/streptomycin (5000 units/mL penicillin, 5000 µg/mL streptomycin), 2 mM L-glutamine (200 mM) and 0.1 mM 2-mercaptoethanol (50 mM)). Tissue samples obtained from young donor patients were directly minced into explants without collagenase digestion. After 2 to 3 weeks, phase-bright cells growing on top of the cardiac fibroblast outgrowth, were collected and cultured at low density (15,000–30,000 cells/mL) on poly-D-Lysin-coated (Millipore) culture plates in cardiosphere-growing medium (CGM) (35% IMDM and 65% D-MEM-F12 supplemented with 10% FBS, 1% penicillin/streptomycin (5000 units/mL penicillin, 5000 µg/mL streptomycin), 2 mM L-glutamine (200 mM), 0.1 mM 2-mercaptoethanol (50 mM) and 1X B-27 supplement (50X)). After ca. 5 days, cardiospheres were collected and plated on fibronectin-coated (25 µg/mL) culture plates in CGM medium. Phase-bright cells were harvested from individual explant cultures every 3 to 5 days up to 4 consecutive times. Medium of CDCs was changed twice a week and cells were passaged when 90% confluency was reached.
Sequencing and data analysis
Five patient samples per group were included in the sequencing analysis (c-kitpos yCPC: yCPC11, yCPC12, yCPC13, yCPC14, yCPC15; c-kitpos aCPC: aCPC13, aCPC14, aCPC15, aCPC16, aCPC17; yCDC: yCDC2, yCDC3, yCDC4, yCDC6, yCDC7; aCDC: aCDC1, aCDC2, aCDC4, aCDC5, aCDC6). Total RNA was extracted using the Qiagen RNeasy micro kit with DNase treatment, followed by a clean-up step with the Qiagen RNeasy minElute columns. Quality control was performed using the Nanodrop Spectrophotometer (A260/280 > 1.8 and A260/230 > 1.5) and Agilent Bioanalyzer (RIN-value > 7). Each sample was sequenced individually on the NextSeq. 500 (High Output, 75 bp, SR; Illumina) using the Illumina TruSeq® Stranded mRNA Sample Prep Kit and 250 ng as input RNA. Reads were pre-processed accordingly and aligned with Tophat v2.0.13 to the reference genome of Homo Sapiens (GRCh37.73). The number of reads in the alignments that overlap with gene features were counted with featureCounts 1.4.6. Raw counts were further corrected within samples for GC-content and between samples using full quantile normalization, as implemented in the EDASeq package from Bioconductor. The Spearman correlation was calculated between all samples using the normalized counts as expression values.
With the EdgeR 3.8.6 package of Bioconductor, a negative binomial generalized linear model (GLM) was fitted against the normalized counts. Differential expression was calculated with a GLM likelihood ratio test in EdgeR. The resulting P-values were corrected for multiple testing with Benjamini-Hochberg (FDR, False Discovery Rate). Differential expression was calculated between aCDCs versus yCDCs and between aCDCs and c-kitpos aCPCs. Genes with a FDR P < 0.05 and a fold change of |1| or more was considered significant.
String network analysis was performed on the top significant genes37. Clustering on the network analysis was performed using the MCL algorithm with inflation parameter set to 3. Gene ontology analysis to identify underlying biological processes was performed using DAVID Functional Annotation Tool 6.838. All differentially expressed genes were submitted to Ingenuity Pathway Analysis (IPA). The top canonical pathways, significantly enriched by the differentially expressed genes, were selected by performing a Fisher Exact t-test (P < 0.05) on the ratio of enriched genes divided by the total number of genes in the pathway. A z-score statistic was calculated to predict the activation state of the pathway based on the fold change of the genes included in that pathway, with a cut-off z-score of >|1.5|. The sequencing data has been made available through the GEO website.
Real-time qRT-PCR
Total RNA from cultured cells was isolated using RNeasy Micro Kits (Qiagen) with DNase treatment. The first strand reverse transcription was performed using GoScript Reverse Transcription System (Promega). Real-time qRT-PCR was performed using TaqMan Fast Universal PCR Master Mix and TaqMan Gene Expression Assays on a StepOnePlus System (Applied Biosystems). All reactions were run in duplicate and results were expressed as 2−ΔΔCt. Relative gene expression was normalized to the housekeeping genes ACTB and GAPDH and an interplate calibrator was used to compare results between plates.
Flow cytometry analysis
CDCs and c-kitpos CPCs were trypsinized at passage 5 to 7 and 50,000–100,000 cells were incubated with fluorochrome-conjugated antibodies (Table S1). Samples were evaluated on a FACSCanto II flow cytometer (BD Biosciences) with at least 10,000 recorded events. Cellular debris and aggregates were gated out based on forward and side scatter. Data were analysed with FACSDiVa software (BD Biosciences).
In vitro proliferation potential
Growth kinetics were determined by calculating cell numbers at every passage to derive expansion curves and PDT throughout the entire expansion process. PDT was computed by linear regression of log2 values of cell number. Only values in the exponential growth phase were used.
In vitro vasculogenic potential
Tube formation assay was performed as described previously39. Briefly, 65,000 c-kitpos CPCs or CDCs per well were plated on 24-well plates coated with Matrigel® basement membrane matrix (BD Biosciences). Tube formation analysis was performed after 6 hours of incubation through phase-contrast mosaic images (Axiovert 200 M imaging microscope, Carl Zeiss). Angiogenesis analyser software (Gilles Carpentier) for ImageJ (National Institutes of Health) was used for morphometric analysis of the obtained images. Additionally, a 3D sprouting assay in Matrigel® was performed using spheroids as described previously40. Per group, cells of 2 different donors were used and for each donor, 4 different spheroids were imaged. C-kitpos CPC and CDC spheroids containing 5,000 cells were formed in culture medium supplemented with poly(vinyl alcohol) in V-shaped 96-well plates for 96 hours. Formed spheroids were transferred to glass bottom culture plates coated with Matrigel®. A second layer of Matrigel® was added to accomplish a 3D structure. The sprouting was observed after 48 hours incubation through phase-contrast images (Confocal laser scanning microscope LSM 800, Carl Zeiss). To visualize the 3D sprouting, a Z-stack was made.
Immunocytochemical analysis
For immunofluorescence staining, cells were fixed with 4% paraformaldehyde (PFA), permeabilized with 0.2% Triton-X100 and incubated with blocking solution (2% BSA) for one hour at RT. Next, cells were incubated overnight at 4 °C with primary antibodies, washed and incubated with secondary antibodies for 1 hour at RT. Secondary antibodies were conjugated with biotin to allow amplification of the signal (Table S2). For amplification, the tyramide signal amplification kit (TSA, Perkin Elmer) was used. Nuclei were stained with DAPI. Images were obtained using an Axiovert 200 M imaging microscope (Carl Zeiss).
Paracrine assay
To compare the production of growth factors and cytokines, CDCs and c-kitpos CPCs were seeded in 6-well culture plates in PBS with 2% FBS for 24 hours. Conditioned medium was collected and the concentrations of VEGF, bFGF, IGF-1, HGF, PlGF and SDF-1 were measured using human-specific ELISA kits (R&D Systems), according to the manufacturer’s instructions. In addition, total protein levels were determined by a bicinchoninic acid assay (Pierce BCA Protein Assay Kit, Thermo Scientific), according to the manufacturer’s instructions to normalize the production of growth factors and cytokines.
Conditioned medium and exosome purification
CDCs (yCDC, n = 2; aCDC, n = 2) and c-kitpos CPCs (c-kitpos yCPC, n = 3; c-kitpos aCPC, n = 3) were cultured for 48 hours in serum-free medium, before collection conditioned medium. Exosomes were isolated using the ultracentrifugation method. Briefly, cells were pelleted by low speed centrifugation (15 min. at 500 g). The supernatant, containing the exosomes and cell debris, were then centrifuged for 30 min. at 10,000 g to pellet the cell debris. The residual supernatant was then finally ultracentrifuged in 2 different steps (3 hours at 140,000 g, 70 min. at 140,000 g) to obtain the exosome fraction. The purified exosome pellet was dissolved in PBS for further analysis. All centrifugation and ultracentrifugation steps were performed at 4 °C. The nanoparticle analysis technology (Nanosight) was used to measure particle size and concentration. Six technical replicates per exosome sample were performed. The extraction of total RNA of the exosomes was performed using the total exosome RNA and protein isolation kit (Life technologies). The total exosome RNA yield and quality was measured by a spectrophotometer (NanoDrop2000, Thermo scientific). Normalized expression levels of miR-22-3p, miR-132-3p, miR-146a-3p and miR-210-3p were calculated using miR-16 as a housekeeping gene.
In vitro differentiation assays of mono-culture of human stem cells
To induce CM differentiation, c-kitpos CPCs and CDCs were seeded at densities of 5 × 103 cells/cm2. The c-kit expansion or CGM maintenance medium (CON) was replaced by cardiomyocyte differentiation medium (Millipore) (DIFF) for 12 days with fractional medium changes of 50% every 2–3 days. Cells were stained for sarcomeric alpha-actinin and F-actin to determine CM differentiation. Images were obtained using an Axiovert 200 M imaging microscope (Carl Zeiss) and quantification was done using ImageJ software (National Institutes of Health).
Culture conditions SWAP experiment
c-kitpos CPCs were cultured in CDC expansion medium (35% IMDM and 65% D-MEM-F12 supplemented with 10% FBS, 1% penicillin/streptomycin (5000 units/mL penicillin, 5000 µg/mL streptomycin), 2 mM L-glutamine (200 mM), 0.1 mM 2-mercaptoethanol (50 mM) and 1X B-27 supplement (50X)) and fibronectin-coated (25 μg/mL) culture plates and CDCs in c-kitpos CPC expansion medium (Ham’s F12 medium supplemented with 10% FBS, 1% penicillin/streptomycin (5000 units/mL penicillin, 5000 µg/mL streptomycin), 5 mU/mL human erythropoietin (EPO)) for at least 2 passages. Total RNA from cultured cells was isolated and qRT-PCR was performed. In addition, cells were plated on Matrigel® basement membrane matrix-coated culture plates for a tube formation assay.
Statistical analysis
All data are presented as mean ± standard error of the mean (SEM) except for the RT-qPCR results, which are presented as geometric mean with 95% confidence interval (CI). Intergroup differences were analysed using a two-tailed unpaired t-tests or one-way ANOVA followed by a Bonferroni post-hoc test or a chi-square test for normally distributed data. Non-normally distributed data were compared using a non-parametric Mann-Whitney test or non-parametric Kruskal-Wallis test followed by a Dunn’s post-hoc test. Normality of the data was tested using a Shapiro-Wilk normality test. Spearman non-parametric correlation was used to analyse linear co-variation. The goodness of fit was expressed as the coefficient of determination R2. A probability value of P < 0.05 was considered statistically significant. All analyses were performed using Prism 6.0e software (GraphPad Software).
Data availability
The sequencing data generated and analysed during the current study has been made available through the GEO website.
References
Cleland, J. G. & McGowan, J. Heart failure due to ischaemic heart disease: epidemiology, pathophysiology and progression. J Cardiovasc Pharmacol 33(Suppl 3), S17–29 (1999).
White, H. D. et al. Mortality and morbidity remain high despite captopril and/or Valsartan therapy in elderly patients with left ventricular systolic dysfunction, heart failure, or both after acute myocardial infarction: results from the Valsartan in Acute Myocardial Infarction Trial (VALIANT). Circulation 112, 3391–3399, https://doi.org/10.1161/CIRCULATIONAHA.105.551143 (2005).
Menasche, P. Skeletal myoblasts for cardiac repair: Act II? J Am Coll Cardiol 52, 1881–1883, https://doi.org/10.1016/j.jacc.2008.07.066 (2008).
Orlic, D. et al. Bone marrow cells regenerate infarcted myocardium. Nature 410, 701–705, https://doi.org/10.1038/35070587 (2001).
Hare, J. M. et al. Comparison of allogeneic vs autologous bone marrow-derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: the POSEIDON randomized trial. JAMA 308, 2369–2379, https://doi.org/10.1001/jama.2012.25321 (2012).
Bergmann, O. & Jovinge, S. Cardiac regeneration in vivo: mending the heart from within? Stem Cell Res 13, 523–531, https://doi.org/10.1016/j.scr.2014.07.002 (2014).
Beltrami, A. P. et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114, 763–776 (2003).
Matsuura, K. et al. Adult cardiac Sca-1-positive cells differentiate into beating cardiomyocytes. J Biol Chem 279, 11384–11391, https://doi.org/10.1074/jbc.M310822200 (2004).
Pfister, O. et al. Role of the ATP-binding cassette transporter Abcg2 in the phenotype and function of cardiac side population cells. Circ Res 103, 825–835, https://doi.org/10.1161/CIRCRESAHA.108.174615 (2008).
Smith, R. R. et al. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 115, 896–908, https://doi.org/10.1161/CIRCULATIONAHA.106.655209 (2007).
Dey, D. et al. Dissecting the molecular relationship among various cardiogenic progenitor cells. Circ Res 112, 1253–1262, https://doi.org/10.1161/CIRCRESAHA.112.300779 (2013).
Bolli, R. et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet 378, 1847–1857, https://doi.org/10.1016/S0140-6736(11)61590-0 (2011).
Malliaras, K. et al. Intracoronary cardiosphere-derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dySfunction). J Am Coll Cardiol 63, 110–122, https://doi.org/10.1016/j.jacc.2013.08.724 (2014).
Chakravarty, T. et al. Abstract 18236: Dilated cardiomYopathy iNtervention With Allogeneic MyocardIally-regenerative Cells (DYNAMIC): Six Month Safety and Exploratory Efficacy Results. Circulation 132, A18236 (2015).
Cheng, K. et al. Relative roles of CD90 and c-kit to the regenerative efficacy of cardiosphere-derived cells in humans and in a mouse model of myocardial infarction. J Am Heart Assoc 3, e001260, https://doi.org/10.1161/JAHA.114.001260 (2014).
Askari, A. T. et al. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet 362, 697–703, https://doi.org/10.1016/S0140-6736(03)14232-8 (2003).
Ibrahim, A. G., Cheng, K. & Marban, E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports 2, 606–619, https://doi.org/10.1016/j.stemcr.2014.04.006 (2014).
Porrello, E. R. et al. Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080, https://doi.org/10.1126/science.1200708 (2011).
Liu, Z. et al. ENDOGLIN is dispensable for vasculogenesis, but required for vascular endothelial growth factor-induced angiogenesis. PLoS One 9, e86273, https://doi.org/10.1371/journal.pone.0086273 (2014).
Dominici, M. et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317, https://doi.org/10.1080/14653240600855905 (2006).
Buckingham, M., Meilhac, S. & Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet 6, 826–835, https://doi.org/10.1038/nrg1710 (2005).
Fuentes, T. I. et al. Human neonatal cardiovascular progenitors: unlocking the secret to regenerative ability. PLoS One 8, e77464, https://doi.org/10.1371/journal.pone.0077464 (2013).
Nakamura, T. et al. Influence of aging on the quantity and quality of human cardiac stem cells. Sci Rep 6, 22781, https://doi.org/10.1038/srep22781 (2016).
Simpson, D. L. et al. A strong regenerative ability of cardiac stem cells derived from neonatal hearts. Circulation 126, S46–53, https://doi.org/10.1161/CIRCULATIONAHA.111.084699 (2012).
Sharma, S. et al. A Deep Proteome Analysis Identifies the Complete Secretome as the Functional Unit of Human Cardiac Progenitor Cells. Circ Res 120, 816–834, https://doi.org/10.1161/CIRCRESAHA.116.309782 (2017).
Mishra, R. et al. Characterization and functionality of cardiac progenitor cells in congenital heart patients. Circulation 123, 364–373, https://doi.org/10.1161/CIRCULATIONAHA.110.971622 (2011).
Chimenti, I. et al. Relative roles of direct regeneration versus paracrine effects of human cardiosphere-derived cells transplanted into infarcted mice. Circ Res 106, 971–980, https://doi.org/10.1161/CIRCRESAHA.109.210682 (2010).
Johnstone, R. M. Revisiting the road to the discovery of exosomes. Blood Cells Mol Dis 34, 214–219, https://doi.org/10.1016/j.bcmd.2005.03.002 (2005).
Barile, L. et al. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc Res 103, 530–541, https://doi.org/10.1093/cvr/cvu167 (2014).
Aminzadeh, M. A. et al. Therapeutic efficacy of cardiosphere-derived cells in a transgenic mouse model of non-ischaemic dilated cardiomyopathy. Eur Heart J 36, 751–762, https://doi.org/10.1093/eurheartj/ehu196 (2015).
de Couto, G. et al. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Invest 125, 3147–3162, https://doi.org/10.1172/JCI81321 (2015).
de Couto, G. et al. Exosomal MicroRNA Transfer Into Macrophages Mediates Cellular Postconditioning. Circulation 136, 200–214, https://doi.org/10.1161/CIRCULATIONAHA.116.024590 (2017).
Frangogiannis, N. G. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 11, 255–265, https://doi.org/10.1038/nrcardio.2014.28 (2014).
Frangogiannis, N. G., Smith, C. W. & Entman, M. L. The inflammatory response in myocardial infarction. Cardiovasc Res 53, 31–47 (2002).
Frangogiannis, N. G. Regulation of the inflammatory response in cardiac repair. Circ Res 110, 159–173, https://doi.org/10.1161/CIRCRESAHA.111.243162 (2012).
Darwich, L. et al. Secretion of interferon-gamma by human macrophages demonstrated at the single-cell level after costimulation with interleukin (IL)-12 plus IL-18. Immunology 126, 386–393, https://doi.org/10.1111/j.1365-2567.2008.02905.x (2009).
Szklarczyk, D. et al. STRINGv10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–452, https://doi.org/10.1093/nar/gku1003 (2015).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57, https://doi.org/10.1038/nprot.2008.211 (2009).
Dauwe, D. et al. Neovascularization Potential of Blood Outgrowth Endothelial Cells From Patients With Stable Ischemic Heart Failure Is Preserved. J Am Heart Assoc 5, e002288, https://doi.org/10.1161/JAHA.115.002288 (2016).
Korff, T. & Augustin, H. G. Tensional forces in fibrillar extracellular matrices control directional capillary sprouting. J Cell Sci 112(Pt 19), 3249–3258 (1999).
Acknowledgements
Grant support PF-KUL (#PF-2010/014 to SJ) & EU-FP7 CARE-MI (#242038 to SJ/MS).
Author information
Authors and Affiliations
Contributions
A.S.W., M.V., M.S., A.L. and S.J. wrote the main manuscript text. A.S.W. prepared Figures 1–5 and supplemental data, M.V. contributed to Figure 2. H.G. contributed to Figures 3–5. L.O. and N.V.D. contributed to Figure 5. B.M., P.H., F.R. and R.H. provided the human tissue biopsies. S.T. obtained written informed consent from the patients. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Walravens, AS., Vanhaverbeke, M., Ottaviani, L. et al. Molecular signature of progenitor cells isolated from young and adult human hearts. Sci Rep 8, 9266 (2018). https://doi.org/10.1038/s41598-018-26969-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-26969-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
