
Abstract
As aging involves oxidant injury, we examined the role of the recently described Na/K-ATPase oxidant amplification loop (NKAL). First, C57Bl6 old mice were given a western diet to stimulate oxidant injury or pNaKtide to antagonize the NKAL. The western diet accelerated functional and morphological evidence for aging whereas pNaKtide attenuated these changes. Next, human dermal fibroblasts (HDFs) were exposed to different types of oxidant stress in vitro each of which increased expression of senescence markers, cell-injury, and apoptosis as well as stimulated the NKAL. Further stimulation of the NKAL with ouabain augmented cellular senescence whereas treatment with pNaKtide attenuated it. Although N-Acetyl Cysteine and Vitamin E also ameliorated overall oxidant stress to a similar degree as pNaKtide, the pNaKtide produced protection against senescence that was substantially greater than that seen with either antioxidant. In particular, pNaKtide appeared to specifically ameliorate nuclear oxidant stress to a greater degree. These data demonstrate that the NKAL is intimately involved in the aging process and may serve as a target for anti-aging interventions.
Similar content being viewed by others
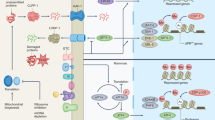


Introduction
Aging is characterized by a number of physiological changes including loss of cell division, oxidative stress, DNA damage, nuclear changes, and increased expression of senescence-associated genes1,2,3,4,5. From the phenotypic point of view, aging might be defined as the progressive age-related decline of physiological function caused by cell arrest (senescence) and/or programmed cell death (apoptosis)6,7. It has been known for some time that oxidant stress plays a central role in the aging process, and is causally involved in the injury to cellular proteins and DNA8,9,10. When reactive oxygen species (ROS) accumulation exceeds the detoxifying ability of the cell, the resulting oxidative stress induces damage, senescence, and apoptosis. It is the imbalance between ROS and antioxidant defense mechanisms11 that contribute to impaired physiological function, disease development, and ultimately, the limited life span of an organism12.
We recently reported that the Na/K-ATPase – Src – EGFR signaling pathway serves as a feed-forward amplification loop for oxidants (Na/K-ATPase oxidant amplification loop, NKAL)13,14,15. Na/K-ATPase can act as a specific receptor for cardiotonic steroids (CTS) and as a non-specific receptor for ROS, inducing conformational changes in Na/K-ATPase α-1 subunit, which in turn, phosphorylates Src, followed by the transactivation of EGFR. This initiates a signaling cascade resulting in additional ROS generation. We further showed that this NKAL is involved in various disease models ranging from uremic cardiomyopathy to obesity13,14,16,17. Our group developed a peptide, pNaKtide, from the N domain of the Na/K-ATPase α1 subunit. This peptide binds Src kinase; ultimately inhibiting the Na/K-ATPase feed forward amplification of ROS16,18,19,20. Based on these earlier observations, we hypothesized that the NKAL might play a role in the aging process and antagonism of this pathway by pNaKtide might attenuate the aging process.
Results
Effect of pNaKtide on body weight, tissue weight, energy expenditure, locomotor activity, and oxygen consumption in C57B16 aging mice
We evaluated the effects of Na/K-ATPase signaling and pNaKtide in vivo using a mouse model of aging and a western diet (WD) regimen to induce oxidative stress. Our results showed that body weight, visceral fat, and subcutaneous fat weights were increased in the old mice and further increased in old mice fed a WD (Table 1). These increases were significantly decreased by pNaKtide treatment (Table 1). There were no significant differences in any of these measures between young mice and young mice treated with pNaKtide.
There were no significant differences in heart weight between any of the groups (Table 1). Energy expenditure was determined as heat production in kcal/kg/hr21. The results showed that there was a significant increase in energy expenditure in pNaKtide treated old mice fed a WD as compared to old mice fed a WD (Table 1). Locomotor activity was also measured based on the ambulatory count over a 24 hr period21. Old mice showed decreased activity as compared to young mice, and old mice fed a WD showed decreased activity as compared to young mice, old baseline, and old mice. Administration of pNaKtide to old mice significantly increased their levels of locomotor activity compared to old baseline and old mice. Administration of pNaKtide to old mice fed a WD significantly increased their levels of locomotor activity compared to old mice fed a WD (Table 1). Oxygen consumption was measured in terms of mL/kg/hr. The results showed that there was a significant increase in oxygen consumption in pNaKtide treated old mice fed a WD as compared to old mice fed a WD (Table 1).
Effect of pNaKtide on adipocyte phenotype, senescence, and Na/K-ATPase signaling in C57Bl6 aging mice
Adipose tissue dysfunction occurs with aging22,23. Hematoxylin and eosin (H&E) staining of visceral fat tissues showed increased adipocyte size in adipose tissue of old mice with further increases in old mice fed a WD (Fig. 1A and B). Crown-like structures, representing an accumulation of macrophages around adipocytes, were apparent in adipose tissues of old mice with further increases noted in old mice fed a WD (Fig. 1A). In both groups, these increases were significantly attenuated with pNaKtide treatment. The TUNEL assay demonstrated that apoptosis was significantly increased in the adipose tissue of old mice and further increased in old mice fed a WD. These increases were also attenuated with pNaKtide treatment (Fig. 1C). The expression level of peroxisome proliferator-activated receptor gamma (PPAR-γ) is upregulated in diet-induced obesity, indicative of adipocyte hypertrophy and physiologic dysregulation of adipose tissue24,25. Apolipoprotein J (ApoJ) and p21 are known to increase in stress-induced cellular senescence25,26. Our results showed that ApoJ, p21, and PPARγ levels were increased in the old mice and further increased in old mice fed a WD (Fig. 1D,E and F). The magnitudes of these increases were significantly decreased with pNaKtide treatment. Protein carbonylation is well-known indicator of oxidative stress15. Old mice had demonstrable increases in protein carbonylation in adipose tissues, and these increases were amplified with a WD (Fig. S1A). The pSrc levels were also increased in the adipose tissues of old mice with further increases with a WD (Fig. S1B). Again, these changes were negated by treatment with pNaKtide. Levels of TNFα, a pro-inflammatory cytokine, and thiobarbituric reactive substances (TBARS), another marker of oxidative stress, increased significantly in old mice and old mice fed a WD as compared to the young mice (Fig. S1C and D). These increases were again negated by treatment with pNaKtide.

Effects of pNaKtide on adipocyte phenotype, senescence, and apoptosis in C57Bl6 aging mice. (A) H&E staining in visceral adipose tissue. Images taken with 40X objective lens; scale bar represents 100 µm. Arrows mark “crown like structures” indicative of inflammation. (B) Quantitative analysis of adipocytes area in visceral adipose tissue. (C) Representative images of TUNEL assay with quantification in C57B16 aging mice. Images taken with 40X objective lens; scale bar represents 25 µm. (D–F) qRT-PCR analysis of ApoJ, p21 and PPARγ in C57Bl6 aging mice with GAPDH as a loading control. Y, young; Y + P, young + pNaKtide; OB, old baseline; O, old; O + P, old + pNaKtide; O + WD, old + western diet; O + WD + P, old + western diet + pNaKtide. N = 8/group, *p < 0.05, **p < 0.01 vs Y, #p < 0.05, ##p < 0.01 vs O, &p < 0.05, &&p < 0.01 vs O + WD.
Effect of pNaKtide on heart senescence, apoptosis, and Na/K-ATPase signaling in C57Bl6 aging mice
Sirius red staining was performed to quantify fibrosis in heart tissues. Sirius red staining demonstrated a significant increase in fibrosis of the heart tissue in the old mice with further increases in old mice fed a WD (Fig. 2A and B). Treatment with pNaKtide decreased the fibrosis in both of these groups. H&E staining of cardiac tissues demonstrated remnants of degenerating myofibers in the old mice and old mice fed a WD (data not shown). Again, these changes were attenuated by pNaKtide treatment.

Effect of pNaKtide on fibrosis, senescence markers, and apoptosis in heart tissue of C57Bl6 aging mice. (A and B) Sirius red staining for detection of cardiac fibrosis with quantitative analysis. Images taken with 20X objective lens; scale bar represents 100 µm. (C–E) qRT-PCR analysis of Fibronectin, p21 and ApoJ in C57B16 aging mice. GAPDH was used as a loading control. (F,G) Representative images of TUNEL assay with quantification in C57B16 aging mice. Images taken with 40X objective lens; scale bar represents 25 µm. N = 8/group, *p < 0.05, **p < 0.01 vs Y, #p < 0.05, ##p < 0.01 vs O, &p < 0.05, &&p < 0.01 vs O + WD.
Aging resulted in cardiac hypertrophy and diastolic dysfunction as assessed by echocardiographic methods, summarized in Table 2. Specifically, old mice and old mice fed a WD had increased left ventricular wall thickness (anterior wall thickness (AWT), posterior wall thickness (PWT), relative wall thickness (RWT)), and left ventricular mass index (LVMI)). In addition, impaired function was noted in these groups with the myocardial performance index (MPI). These changes were significantly attenuated by pNaKtide treatment (Table 2). A significant increase in fibronectin, p21, and ApoJ levels were seen in both the old mice and old mice fed a WD (Fig. 2C,D and E). The magnitudes of these increases were significantly decreased with pNaKtide treatment. The TUNEL assay demonstrated that apoptosis was significantly increased in the hearts of old mice and further increased in old mice fed a WD (Fig. 2F and G). These increases were attenuated with pNaKtide treatment. There was significant increase in protein carbonylation in old mice with further increases in old mice fed a WD. These increases were attenuated with pNaKtide (Fig. S2A). TBARS levels followed a similar trend (Fig. S2B).
Effect of pNaKtide on H2O2-induced senescence in human dermal fibroblasts (HDFs)
Administration of H2O2 results in oxidative stress and DNA damage, which can lead to senescence27,28. Dose concentration curves were generated to determine the optimal dose of pNaKtide, which is effective in attenuating senescence (Fig. S3A). Our results showed that H2O2 treated HDF exhibited morphological changes indicative of cellular senescence that included enlargement, flattening, and elongation of the cells along with decreased cell number, when viewed under light microscopy (Fig. S3A). We also noted that expression of senescence genes, ApoJ, p21, and fibronectin29 exhibited significant upregulation after subjection to H2O2 treatment compared to untreated, control cells (Fig. S3B, S3C, S3D). Treatment with 1 µM pNaKtide significantly attenuated this effect when administered to H2O2 treated cells. Elevated levels of beta-galactosidase (SA β-Gal), senescence-associated marker30, and phosphorylated histone H2AX (γ-H2AX), a marker of double-stranded DNA breaks31, were noted in the H2O2 group and these changes were also attenuated by pNaKtide (Fig. 3A and B). Using a TUNEL assay to detect levels of fragmented and degraded DNA32, we found that apoptosis levels were elevated in cells treated with H2O2, which was attenuated by pNaKtide treatment. Caspase-9 is an essential initiator caspase required for apoptosis signaling33. Caspase-9 activity was elevated in H2O2 treated cells and this was decreased by treatment with pNaKtide (Fig. 3C and D). No significant differences between the control group and the control group treated with pNaKtide were noted.

Effects of pNaKtide on H2O2-induced senescence markers, apoptosis, and Na/K-ATPase signaling in human dermal fibroblasts (HDFs). (A) Qualitative and quantitative analysis of ß-galactosidase levels. (B) Representative images and quantification of γ-H2AX levels. Images taken with 40X objective lens; scale bar represents 100 µm. (C) Representative images and quantification of the TUNEL assay (D) Quantitative analysis of Caspase-9 activity in cell lysates. (E) pATM, (F) pCHK2, and (G) p53 immunoblot analysis. Data shown are the mean band density normalized to ATM, CHK2, and actin, respectively. (H) Protein carbonylation levels with Coomassie staining as a loading control. (I) pSrc immunoblot analysis with data shown as mean band density normalized to Src. Data are displayed as “scatter plots” showing data points and “box plots” showing the distribution of a continuous variable as described in the Methods section. All gels were cropped above and below the band. N = 6/group, *p < 0.05, **p < 0.01 vs. control, #p < 0.05, ##p < 0.01 vs. H2O2.
Effect of pNaKtide on the p53 senescence pathway and Na/K-ATPase signaling in H2O2-treated HDFs
Cell senescence can be induced and maintained through the p53 pathway34,35,36. The effects of oxidative stress, such as double-strand breaks in DNA, trigger activation of ATM and CHK2, which then activates p537,26,37,38,39. Treatment of HDFs with H2O2 significantly increased levels of pATM, pCHK2, and p53 protein compared with untreated controls. This effect was significantly attenuated following pNaKtide treatment (Fig. 3E,F and G). Further, the pNaKtide-treated group had significantly lower p21 mRNA expression compared with the H2O2-treated group (Table 3). Ki-67 is a well-established marker of cell proliferation40,41. The H2O2-treated group had significantly lower expression of Ki-67 mRNA and expression levels were restored in the pNaKtide-treated group (Table 3). Further, mRNA levels of other senescence markers, including ApoJ, matrix metalloproteinase 9 (MMP9), fibronectin, and collagenase were measured. All four were significantly decreased in the pNaKtide-treated group compared with the H2O2-treated group (Table 3). Protein carbonylation increased in H2O2-treated cells, and this increase was negated by pNaKtide treatment (Fig. 3H). Treatment with pNaKtide also blocked Na/K-ATPase-regulated Src activation in H2O2-treated HDFs (Fig. 3I). Lactate dehydrogenase (LDH) release, a marker of cytotoxicity42, was increased in the H2O2-treated group, and this was attenuated by concomitant treatment with pNaKtide (Table 3).
Effect of anti-oxidants on H2O2-induced senescence in HDFs
NAC and Vitamin E are known antioxidants, which have been shown to be effective in attenuating cellular senescence43,44,45,46,47. Dose concentration curves were generated to determine the optimal doses of NAC and Vitamin E, which were somewhat effective in attenuating senescence in our H2O2 model (Fig. 4A). Cells exposed to H2O2 and treated with 5 mM concentration of NAC or 50uM concentration of Vitamin E attenuated the morphological or biochemical changes of senescence, but less so than pNaKtide (Fig. 4A,C–E). However, the reductions in cellular oxidant stress as assessed by TBARS achieved with NAC and Vit E were virtually identical to that seen with pNaKtide (Fig. 4B).

Effect of anti-oxidants on H2O2 induced cellular senescence in HDFs. (A) Representative images of HDFs exposed to H2O2 and treated with varying concentrations of NAC and Vitamin E respectively. Images taken with 20X objective lens; scale bar represents 100 µm. (B) Quantitative analysis of TBARS. (C) Representative images and quantification of γ-H2AX levels. Images taken with 40X objective lens; scale bar represents 100 µm. qRT-PCR analysis of (D) p21 and (E) MMP9 in HDFs exposed to H2O2. GAPDH was used as a loading control. N = 6/group. **p < 0.05 vs. CTR, ##p < 0.01 vs. H2O2, &&p < 0.01 vs. H2O2 + pNaKtide.
Effect of pNaKtide on UV radiation-induced senescence in HDFs
HDFs were treated with UV radiation, another mechanism for triggering oxidative-stress–mediated senescence48. Levels of SA β-Gal and TBARS were significantly higher in the UV-treated group compared with the control group (Fig. S4A and B). These increases were attenuated in the pNaKtide-treated group. The mRNA expression levels of p21, ApoJ, MMP9, and fibronectin were all significantly higher in the UV-treated group, and these increases were negated when cells were also treated with pNaKtide (Fig. S4C, S4D, S4E, and S4F).
Effect of pNaKtide on ouabain-induced senescence in HDFs
Since H2O2-mediated ROS initiated Na/K-ATPase signaling and induced senescence, we next studied whether ouabain, a specific receptor for Na/K-ATPase signaling activation, could also induce cellular senescence. A concentration curve was generated to determine the optimal dose of ouabain, and it was found that treatment with a 50 nM concentration significantly exhibited morphological changes indicative of cellular senescence (Fig. S5A). Additionally, LDH release increased significantly in the ouabain-treated group compared with the control group (Fig. S6B). Our results showed a significant increase in levels of γ-H2AX (Fig. S5C), and pATM, pCHK2, p53 protein (Fig. S6A,B and C) in the ouabain treated group that were attenuated with pNaKtide treatment. Also, pNaKtide decreased p21 mRNA expression when administered with ouabain (Fig. S6D). Caspase 9 activity increased significantly in the ouabain-treated group; this was also significantly attenuated by concomitant treatment with pNaKtide (Fig. S5D). The ouabain treated group also had significantly decreased Ki-67 levels as compared to the control group. This decrease was negated with pNaKtide treatment (Fig. S6E). Ouabain, of course, increased the pSrc/Src ratio, and this was also significantly decreased by pNaKtide treatment (Fig. S6F).
Effect of glucose oxidase (GO) and pNaKtide on oxidative stress, cell proliferation, and apoptosis in HDFs
GO generates moderate amounts of H2O2 which results in a steady increase in intracellular ROS in HDFs49. Concentrations of GO ranging from 0 to 10 mU/ml were administered to HDFs. Our result showed that TBARS level increased with increasing concentrations of GO (Fig. 5A). Treatment with pNaKtide attenuated this increase; however it is important to note that TBARS concentration in the pNaKtide treated cells was still demonstrably elevated with the high concentrations of GO. To determine the role of Na/K-ATPase signaling in maintaining cell function and proliferation, MTT and CyQUANT assays were performed. Based on the MTT assay, 1 and 3 mU/ml of GO increased cell proliferation compared with control group, whereas 5 and 10 mU/ml of GO decreased cell count (Fig. 5B). Based on these observations, we hypothesized that some activation of the NKAL, demonstrated with 1 and 3 mU/ml GO may actually result in enhanced cell proliferation. Along these lines, we saw that concomitant treatment with pNaKtide allowed for enhanced cell proliferation in the groups exposed to 5 and 10 mU/ml of GO. The CyQUANT assay showed that compared with controls, the number of cells increased with GO 1 mU/ml and GO 3 mU/ml respectively; whereas 5 and 10 mU/ml of GO decreased cell count (Fig. 5C). Our results further showed that pNaKtide treatment did not increase cell proliferation in the groups exposed to 1, 3, and 5 mU/ml of GO. However, cell counts were increased in cells treated with 10 mU/ml GO with pNaKtide compared with 10 mU/ml GO alone. Based on caspase 9 activity, 5 and 10 mU/ml of GO increased apoptosis significantly compared with the control group. Treatment with pNaKtide negated this effect (Fig. 5D). These results support the concept that oxidants at low concentrations can stimulate cell proliferation whereas at higher concentrations, cell apoptosis results50. Interestingly, pNaKtide not only decreased the net amount of oxidant stress as assessed by the accumulation of TBARS but had more profound effects on cell proliferation and apoptosis than one would anticipate from the antioxidant effect alone.

Effects of glucose oxidase (GO) and pNaKtide on oxidative stress, cell proliferation, and apoptosis in HDFs. Concentrations of GO ranging from 0 to 10 mU/ml were administered to HDFs. (A) Quantitative analysis of TBARS (B) Quantitative analysis of the MTT assay (C) Cell count by CyQUANT assay (D) Caspase-9 activity assay. N = 16/group, *p < 0.05, **p < 0.01 vs. corresponding GO concentration groups without pNaKtide.
Effect of GO and pNaKtide on protein carbonylation in HDFs
Immunofluorescence of protein carbonylation using 2,4-dinitrophenylhydrazine (DNP) showed that 3, 5, and 10 mU/ml of GO significantly increased evidence for carbonylation, more so in the nucleus, compared with the control group (Fig. 6A and B). Furthermore, treatment with pNaKtide significantly reduced the carbonylation levels, especially in the nucleus, compared with GO alone. Immunoblotting of whole cell lysates with anti-DNP antibody showed that 10 mU/ml of GO significantly increased protein carbonylation compared with the control group (Fig. 6C). Conversely, this effect was negated when cells were treated with pNaKtide. Further, a double-staining immunofluorescence study was performed to determine the co-occurrence of protein carbonylation and DNA damage and fluorescent probes for DNP and γ-H2AX were visualized with confocal microscopy (Fig. 6D). HDFs exposed to GO 10 mU/ml concentration showed increased DNA damage along with increased intra-nuclear carbonylation when compared to control showing a positive co-relation between protein carbonylation and DNA damage (Fig. 6E). These increases with GO were attenuated with concomitant pNaKtide treatment.

Effects of pNaKtide and GO on protein carbonylation in HDFs. (A) Representative immunofluorescence images of carbonylated protein. HDFs exposed to various concentrations of GO (1 mU, 3 mU, 5 mU, and 10 mU) with or without pNaKtide treatment. DNP fluorescence intensity was visualized using an EVOS portable microscope with an excitation/emission wavelength of 470/525 nm. (B) Quantification was done by Image J software based on the mean optical density transmitted under fluorescent microscopy. Pictures were taken at 40X with a linear scale bar representation of 50 µm. N = 8/group, **p < 0.01 vs. corresponding GO concentration groups without pNaKtide. (C) Representative immunoblot analysis with Coomassie staining as a loading control and the quantification of protein carbonylation levels in HDFs treated with 10 mU/ml GO and pNaKtide. N = 8/group, **p < 0.01 vs. control, #p < 0.01 vs. GO. (D) Confocal images of protein carbonylation and γ-H2AX phosphorylation showing co-occurence of these markers within the nucleus. Nuclei were identified using DAPI staining. DNP and γ-H2A.X double immuno-fluorescence staining were visualized with a Leica SP5 TCSII with Coherent Chameleon multiphoton (MP) Vision II (IR) laser confocal microscope. Pictures were taken with the 100 × objective lens under oil immersion; scale bar representation of 5 µm. N = 8/group. (E) Quantification of DNP intensity and γ-H2AX foci shown as a scatterplot with linear regression demonstrating a strong correlation (R2 = 0.847). Gray band around regression line represent 95% confidence interval.
Discussion
We observed that the recently described NKAL14,15,21 regulates the aging process. We previously showed that a western diet induced Na/K-ATPase signaling and increased oxidative stress in mice14. We used this dietary approach to investigate the effects of age and oxidative stress in adipose tissue, and heart, which are both affected by the aging processes. In both of these organ systems, old mice and old mice fed a WD had evidence for oxidant injury, which was related to the stimulation or inhibition of the NKAL with the WD or pNaKtide, respectively. As in obesity, aging is frequently associated with an increased proportion of adipose tissue as well as increased concentrations of circulating pro-inflammatory cytokines such as TNFα23. In our experiments, old mice had increased fat deposition along with large adipocytes and increased TNFα levels; these changes were accentuated in the old mice fed a WD. Aging of heart tissues is associated with impaired function detectable with echocardiography and fibrosis measurable with histology51. These changes were exacerbated by the WD and attenuated by pNaKtide treatment as well. Again, these changes in adipose tissues were negated with pNaKtide treatment.
HDFs treated with sub-lethal doses of H2O2, UV radiation, ouabain, and GO displayed a senescent phenotype that was attenuated when pNaKtide was added. Treatment of pNaKtide in HDFs exposed to H2O2 and ouabain, respectively, also reduced expression levels of numerous molecular markers of senescence, including pATM, pCHK2, p53, p21, ApoJ, fibronectin, γ-H2AX, SA β-Gal, collagenase, and MMP930,52,53,54,55,56. Inhibition of the NKAL with pNaKtide attenuated oxidation-induced senescence at both morphological and molecular levels, and these improvements were greater than that achieved by the anti-oxidants, N-Acetyl Cysteine and Vitamin E even though these latter antioxidants attenuated oxidant stress to virtually identical degrees as pNaKtide.
We also noted that both H2O2 and ouabain exposure increased levels of cell injury and apoptosis markers. Subsequent pNaKtide treatment decreased injury and apoptosis in the HDF cells as well. Perhaps even more interestingly, our oxidation titration experiments in HDF cells exposed to GO suggest that the Na/K-ATPase oxidant amplification loop preferentially “feeds” oxidants into specific intracellular locations, in particular the nucleus. We suggest that this nuclear oxidant stress effects DNA damage, which is the cornerstone of the aging process. We therefore emphasize that interference with the NKAL with pNaKtide achieves a greater anti-aging effect than would be achieved by simple anti-oxidant administration.
There were a number of limitations in this study. The most obvious is that human tissues were limited to that of an in vitro system, the cultured HDFs. In vivo experiments in the mouse allow for rapid study of processes that take decades in humans, but of course these processes therefore have inherent differences. While cultured cells are a good way to control molecular conditions in aging, this approach has obvious limitations. We would stress however that with the in vitro systems, we were able to identify discordance between the anti-oxidant effects of pNaKtide and pNaKtide’s modulation of cell proliferation and apoptosis, both of which were out of proportion to the anti-oxidant effects. Ligand dependent Na/K-ATPase/Src signaling and transactivation of EGFR has been documented to activate downstream signaling cascades causing ROS generation57,58. Therefore, pharmacological alterations to EGFR may ameliorate cellular oxidative stress and senescence, which might mimic the effects of pNaKtide. Studies are required to investigate the extent of EGFR involvement in the process of aging and cellular senescence, allowing comparing the effectiveness of EGFR silencing with pNaKtide.
Several pharmacological interventions have been developed against aging in an effort to attenuate cellular senescence. These therapeutic drugs eliminate senescent cells by directly targeting the proliferative, apoptotic and cell survival signaling pathways59. The success of targeted anti-aging therapies have been based on a variety of signal cascades including senescence associated secretory phenotype (SASP) modulation (e.g., with resveratrol, apinegin and wogonin), immuno-therapeutics (e.g., anti-IL1R) as well as induction of apoptosis (e.g., quercetin, navitoclax, dasatinib)60,61. In our study, we demonstrated that the Na/K-ATPase oxidant amplification loop plays a vital role in the aging process. Our peptide, pNaKtide, is a specific antagonist of this signaling pathway. The success of this strategy opens up a new approach to the aging process. In the future studies, we plan to compare the anti-aging effects of pNaKtide with other agents like those described above.
Our data clearly suggest that the NKAL is involved in the aging process and, if confirmed in human studies, might ultimately serve as a therapeutic target. If the pNaKtide can be safely used in humans, it might be possible to study the applicability of that specific agent to the problem of clinical aging.
Methods
Experimental design for in vivo experiment
All animal studies were approved by the Marshall University Animal Care and Use Committee in accordance with the National Institutes of Health Guidelines for Care and Use of Laboratory Animals. C57Bl6 young mice (8 weeks old, male) and Old mice (16 months old, male) were purchased from Jackson Laboratory. Upon arrival to the Byrd Biotechnology Center, ARF, Animal Research Facility, mice were placed in cages and were fed normal chow diet and had access to water ad libitum or were fed Western Diet (WD) and had ad libitum access to high fructose solution21. WD containing fructose is a well known model of diet induced metabolic and redox imbalance62. WD contained 42% fat, 42.7% carbohydrate, and 15.2% protein yielding 4.5 KJ/g. Fructose was made at a concentration of 42 g/L, yielding 0.168 KJ/mL. The animals were randomly divided into 7 groups (8–15/group) as follows1: Young2, Old-Baseline3, Old4, Old + WD5, Young + pNaKtide6, Old + pNaKtide, and7 Old + WD + pNaKtide. The number of animals in each group was determined by power analysis following assumptions derived from our previous study with mice63, using a variance of 0.2 within the groups, power of 0.80, and alpha error of 0.05. Old baseline group (16 months) was sacrificed immediately and tissues were saved in liquid nitrogen until assayed. After 4 weeks of control or WD diet respectively, groups 5, 6 and 7 were injected with pNaKtide for 8 weeks (dissolved in saline and injected I.P. at a dose of 25-mg/kg-body weight every 7 days as previously described14). The body weight was measured every week. Young (4 months) and Old (19 months) mice were sacrificed, followed by heart and adipose tissue collection. The tissues were flash frozen in liquid nitrogen and maintained at −80 °C until assayed.
TBARS measurement in adipose and cardiac tissues and HDFs
Oxidative injury in HDFs, adipose tissue, and cardiac tissues were measured using a TBARS assay kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer’s protocol21. Data were normalized to total protein and presented as micromoles per milligram of protein.
Histopathological examination of cardiac and adipose tissue
Heart and visceral fat samples from each group were fixed in 4% paraformaldehyde, dehydrated, embedded in paraffin, and sectioned. Formalin-fixed, paraffin-embedded sections were cut (6 µm thick) and mounted on glass slides. The sections were deparaffinized in xylene. Sections of heart and visceral fat were stained with H&E. Sirius red staining was performed on sections of heart tissue for histological analysis of fibrosis. Preparation, fixation, and data analysis of cardiac and adipose tissue was conducted as previously described64.
Transthoracic Echocardiography
Transthoracic echocardiography (TTE) was performed 24 hours prior to sacrifice as described previously13. The following cardiac characteristics were calculated from the data: myocardial performance index56 = (IVCT + IVRT)/ET, relative wall thickness (RWT) = (PWT + AWT)/EDD, cardiac output (CO) = stroke volume65 × HR/1000, fractional shortening28 = (EDD − ESD)/EDD, ejection fraction (EF) = (EDV − ESV)/EDV and the left ventricular mass index (LVMI) = 1.05[(EDD + PWT + AWT)3 − EDD3]/ body weight (g)13.
Experimental design for in vitro experiments
Primary HDFs were obtained from ATCC (PCS-201-012) and cultured in fibroblast basal media (PCS-201-030) supplemented with low serum growth kit (PCS-201-041). Cultures were maintained at 37 °C in a 5% CO2 incubator and medium was replaced every 48 h. At 80% confluence, the cells were detached using trypsin and split in a 1:3 ratio. HDF cells were plated at a density of 10,000 cells/cm2 for subsequent experiments.
Effect of H2O2, ouabain, UV radiation, GO treatment, NAC, and Vitamin E (α-tocopherol) on cellular senescence
After 24 hours of plating, the cells were treated with 200 µM H2O2 for 2 hours. After 2 hours, they were briefly rinsed with PBS and replaced with normal media with or without pNaKtide at different concentrations ranging from 0.5 µM to 4 µM. Following the dose response experiments, the subsequent treatment with pNaKtide was done at a concentration of 1 µM for 72 hours. For ouabain treatment, after 24 hours of plating, the HDFs were treated with ouabain at different concentrations ranging from 1 nM to 100 nM. Following the dose response experiments, the subsequent treatment with ouabain was done at a concentration of 50 nM with and without 1 µM of pNaKtide for 72 hours. Similarly for UV radiation treatment, after 24 hours of plating, the HDFs were exposed with UV radiation (KERNEL KN-4003 BL) at a distance of 30 cm to the cells through a thin layer of phosphate buffer saline pH 7.4 at room temperature. The cells were treated twice daily for 5 days with UVB at 311 nm for duration of 2 minutes at a dose of 2.5 J/cm2. After every radiation, PBS was replaced with normal media with or without pNaKtide at 1 µM concentration. For the GO experiments, HDF cells were plated 24 hours prior to exposure with an increasing dose of GO (Sigma G-2133) ranging from 1, 3, 5 and 10 mU/ml for 72 hours treated with or without 1 µM pNaKtide. For the treatment with NAC, the HDFs were treated at different concentrations ranging from 1 mM, 5 mM and 10 mM for 72 hours, to generate a dose concentration curve. The subsequent treatment with NAC was done at a concentration of 5 mM. For Vit E treatment, the HDFs were plated for 24 hours and then treated with α-tocopherol at different concentrations ranging from 25 µM to 100 µM for 72 hours. Following the dose response experiments, the subsequent treatment with α-tocopherol was done at a concentration of 50 µM.
Cell viability test by lactate dehydrogenase (LDH) assay
An LDH viability assay kit (Cayman, US) was performed according to the manufacturer’s protocol. HDFs were plated in 96-well plates, and cultured for 24 hours. In the first set of experiments, cells were treated with H2O2 with or without subsequent pNaKtide treatment as described above. In the second set of experiments, HDFs were exposed to ouabain with or without subsequent pNaKtide treatment as described above. The percentage of LDH release for each sample was normalized to the absorbance of samples treated with 0.5% Triton X-100.
Senescence-associated β-galactosidase (SA- β-gal) activity
HDFs were plated in a 6-well plate and exposed to H2O2 or UV radiation with or without subsequent pNaKtide treatment as described above. SA-β-gal activity was detected using a Senescence β-Galactosidase Staining kit (Cell signaling Technology #9860) according to the manufacturer’s protocol. Cells were viewed using light microscopy, and the percentage of cells with SA-β-gal activity was calculated from the number of blue-stained cells divided by total number of cells multiplied by 100.
γ-H2AX immuno-fluorescent staining for DNA Damage
DNA damage was measured in HDFs based on immunofluorescent staining of γ-H2AX. Cells were grown in a six-well plate on coverslips pretreated with poly-lysine. Following exposure to H2O2 or ouabain with or without subsequent pNaKtide treatment as described above, cells were washed with PBS and fixed with cold 1:1 methanol:acetone. Cells were permeabilized with two washes of 0.025% Triton in PBS. Cells were blocked using 1% BSA in PBS for 30 minutes. The cells were incubated overnight at 4 °C with primary antibody, Anti- γ-H2AX antibody (phospho S139; diluted 1:100 in 1% BSA in PBS). Slides were washed in 0.025% Triton in PBS, and the secondary antibody (Alexa Fluor 488 diluted 1:1000 in 1% BSA in PBS) was incubated in the dark at room temperature for 1 hour. Cells were washed three times with PBS. Cells were mounted and counterstained with 50 mg/ml DAPI. Images were collected using a digital inverted microscope that spanned the entire area of the cells; the percentage of γ-H2AX-positive cells was calculated from the number of γ-H2AX-positive cells divided by total number of cells multiplied by 100.
MTT and CyQuant assays for cell proliferation
HDFs were plated in 96-well plates and exposed to GO with or without subsequent pNaKtide treatment as described above. A Vybrant MTT Cell Proliferation Assay Kit (V-13154, Invitrogen) was used according to the manufacturer’s protocol14. CyQUANT Cell Proliferation Assay Kit (C7026, Invitrogen) was used for cell count according to the manufacture’s protocol. Fluorescence was measured at excitation/emission wavelengths of 480/520 nm (Spectramax i3x). Fluorescence measurements were converted to cell number based on a standard curve with an r2 >0.99.
Measurement of caspase-9 activity
In vitro analysis of caspase 9 activity in HDFs was performed using the Caspase 9 Assay Kit (ab65608, Abcam, US) according to the manufacture’s protocol. Caspase 9 activity was measured based on the absorbance at 400–405 nm, collected using a Spectramax i3x. Data were compared with the absorbance of control samples.
TUNEL assay for DNA damage detection
DNA double-strand breaks were detected in HDFs and frozen mouse tissues, (heart and adipose tissue) using the Click-iT® Plus TUNEL Assay (Thermo Fisher Scientific Inc., US) according to the manufacturer’s protocol. Specimens were then mounted using VECTASHIELD® mounting medium and counterstained with 50 mg/ml DAPI. Images were collected using a digital inverted microscope that spanned the entire area of the cells and tissues on the cover slip. The percentage of TUNEL-positive cells was calculated based on the number of positively stained cells divided by the total number of cells multiplied by 100.
DNP assay for protein carbonylation detection
A dinitrophenol (DNP) assay was conducted on HDFs exposed to various concentrations of GO (1, 3, 5, and 10 mU/ml) with or without subsequent pNaKtide treatment. Cells were fixed in methacarn, rinsed with PBS, and incubated with 100 μl DNPH (1 mg/ml in 2 N HCl) for 30 min at room temperature. The sections were blocked for 1 hour with 10% normal horse serum, 0.2% Triton X-100 in PBS. Cells were incubated with primary antibody (rabbit anti-DNP diluted 1:500 in 0.2% Triton X-100 in PBS) overnight at 4 °C. Cells were washed with 0.2% Triton X-100 in PBS then incubated with secondary antibody (donkey anti-rabbit conjugated to green-fluorescent Alexa Fluor 488 dye) for 3 hours. Slides were washed with 0.2% Triton X-100 in PBS and rinsed with deionized H2O. Coverslips were mounted to slides with VECTASHIELD® mounting medium containing DAPI66,67,68. DNP fluorescence intensity was visualized using an EVOS portable microscope with an excitation/emission wavelength of 470/525 nm. Digital photographs of five randomly selected regions of each slide were collected. Image analysis was performed using ImageJ to measure the mean optical density of each cell. Statistical analysis was performed using GraphPad Prism 7.02 (GraphPad Software, Inc.). DNP fluorescence was further visualized with a Leica SP5 TCSII with Coherent Chameleon multiphoton. Vision II (IR) laser confocal microscope at 100x laser power and images taken using 100x objective power.
DNP and γ-H2AX double immuno-fluorescent staining
For the in situ detection of DNA damage and nuclear carbonylation a double staining immunofluorescence assay was performed and visualized using confocal microscopy. Cells were treated, fixed, and permeabilized as described above. Cells were then incubated with 100 μl DNPH for 30 min at RT. The cells were then blocked for 1 hour with 10% normal goat serum, 0.2% Triton X-100 in PBS. Cells were incubated with primary antibodies, rabbit anti-DNP (Sigma-Aldrich), diluted 1:500 in 0.2% Triton X-100 in PBS and mouse anti-γH2AX antibody (Molecular Probes, Inc), simultaneously overnight at 4 °C. The following day, goat anti-rabbit Alexa Flour® 488 IgG and goat anti-mouse Alexa Fluor® 568 IgG H&L were applied for detection of the primary rabbit and mouse antibodies, respectively. Coverslips were mounted to slides with VECTASHIELD® mounting medium containing DAPI. DNP and γ-H2A.X double immuno-fluorescence staining were visualized with a Leica SP5 TCSII with Coherent Chameleon multiphoton (MP) Vision II (IR) laser confocal microscope and images taken under oil at 100x objective power. Image analysis was performed using ImageJ to measure the expression of DNP mean optical density and to calculate the number of γ-H2AX-positive cells divided by total number of cells multiplied by 100. The correlation of the data was determined by graphing the scatterplot and using linear regression analysis.
Western blot analysis
HDFs were pulverized with liquid nitrogen and placed in a homogenization buffer. Homogenates were centrifuged, the supernatant isolated, and immunoblotting was performed as previously reported64,69. The supernatant was used for the determination of p5370, pATM/ATM71, and pCHK2/CHK272; and β actin was used to ensure adequate sample loading for all western blots.
Measurement of c-Src phosphorylation and protein carbonylation
Whole cell lysates from HDFs, visceral fat and cardiac tissues were prepared with NP-40 buffer and activation of c-Src was determined as described previously15,73. Western blotting for protein carbonylation was done on whole-cell lysates from HDFs, adipose and cardiac tissues as described previously73. The signal density values of control samples were normalized to 1.0 with Coomassie staining as a loading control.
RNA extraction and real-time PCR
Total RNA was extracted from HDFs, adipose, and heart tissues using RNeasy Protect Mini kit (QIAGEN, Maryland, USA) according to manufacturer’s instructions. Total RNA (1 μg) was transcribed into cDNA using GeneAmp kit (Applied Biosystems, Branchburg, NJ, USA) reverse transcription reagents. Total RNA was analyzed by a quantitative real time polymerase chain reaction. Real-time PCR was performed using SYBR Green PCR Master Mix on a 7500 HT Fast Real-Time PCR System (Applied Biosystems). Specific primers used were p21, ApoJ, fibronectin, MMP9, collagenase, ki67, and GADPH. Each reaction was performed in triplicate. The comparative threshold cycle3 method was used to calculate the fold amplification as specified by the manufacturer. Based on the literature review, GAPDH was identified as a suitable housekeeping gene for our aging model due to less intergroup variation74,75. However, we performed RT-PCR analysis on two well-established housekeeping genes (HPRT and β-Actin) apart from GAPDH, for validation of a suitable internal control gene in our aging study. Our results demonstrated that GAPDH was a more consistent control than the other tested housekeeping genes in the adipose and heart tissue of C57Bl6 mice (Fig. S7). All experimental samples were normalized using GAPDH as an internal control.
Statistical analyses
The plots used were combination of scatter and boxplots. Scatter plots simply show the data points. The boxplot compactly displays the distribution of a continuous variable. It visualizes five summary statistics (the median, two hinges and two whiskers), and all “outlying” points individually76. The boxplot was defined by:
Notchlower: lower edge of notch = median − 1.58 ∗ interquartile range77/sqrt78
Middle median: 50% quantile
Notchupper: upper edge of notch = median + 1.58 ∗ IQR/sqrt(n)
Lower hinge is the 25th percentile
Upper hinge is the 75th percentile
Data were analyzed and presented with the statistical program R using the packages ggplot2 and doBy. Multiple groups of parametric data (passed Shapiro-Wilk normality test) were compared with one way analysis of variance (ANOVA) and individual group means compared with an unpaired t-test employing the Holm correction for multiple comparisons. Nonparametric data were analyzed with the Wilcoxon rank sum test, also employing the Holm correction for multiple comparisons. All data comparisons are presented at the NS, p < 0.05 and p < 0.01 levels.
Change history
07 May 2020
Editor’s Note: The Editors are currently investigating concerns that have been raised relating to the reliability of data presented in this article. Appropriate editorial action will be taken once the investigation is complete.
01 June 2022
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41598-022-13456-y
References
Davidovic, M. Genetic stability: the key to longevity? Medical hypotheses 53, 329–332 (1999).
Risques, R. A. et al. Ulcerative colitis is a disease of accelerated colon aging: evidence from telomere attrition and DNA damage. Gastroenterology 135, 410–418 (2008).
Fraga, M. F. et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the United States of America 102, 10604–10609 (2005).
Baylin, S. B., Herman, J. G., Graff, J. R., Vertino, P. M. & Issa, J. P. Alterations in DNA methylation: a fundamental aspect of neoplasia. Advances in cancer research 72, 141–196 (1998).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Cerella, C., Grandjenette, C., Dicato, M. & Diederich, M. Roles of Apoptosis and Cellular Senescence in Cancer and Aging. Current drug targets 17, 405–415 (2016).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Yao, H. & Rahman, I. Role of histone deacetylase 2 in epigenetics and cellular senescence: implications in lung inflammaging and COPD. Am J Physiol Lung Cell Mol Physiol 303, L557–566 (2012).
Jones, D. P. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol 295, C849–868 (2008).
Liang, F. Q. & Godley, B. F. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res 76, 397–403 (2003).
Abdollahi, M., Moridani, M. Y., Aruoma, O. I. & Mostafalou, S. Oxidative stress in aging. Oxid Med Cell Longev 2014, 876834 (2014).
Kregel, K. C. & Zhang, H. J. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. American journal of physiology. Regulatory, integrative and comparative physiology 292, R18–36 (2007).
Liu, J. et al. Attenuation of Na/K-ATPase Mediated Oxidant Amplification with pNaKtide Ameliorates Experimental Uremic Cardiomyopathy. Sci Rep 6, 34592 (2016).
Sodhi, K. et al. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci Adv 1, e1500781 (2015).
Yan, Y. et al. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J Biol Chem 288, 34249–34258 (2013).
Li, Z. et al. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. The Journal of biological chemistry 286, 32394–32403 (2011).
Liu, J. et al. Effects of cardiac glycosides on sodium pump expression and function in LLC-PK1 and MDCK cells. Kidney Int 62, 2118–2125 (2002).
Li, Z. et al. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J Biol Chem 284, 21066–21076 (2009).
Liu, J., Kennedy, D. J., Yan, Y. & Shapiro, J. I. Reactive Oxygen Species Modulation of Na/K-ATPase Regulates Fibrosis and Renal Proximal Tubular Sodium Handling. International journal of nephrology 2012, 381320 (2012).
Lai, F. et al. Identification of a mutant alpha1 Na/K-ATPase that pumps but is defective in signal transduction. The Journal of biological chemistry 288, 13295–13304 (2013).
Sodhi, K. et al. pNaKtide Attenuates Steatohepatitis and Atherosclerosis by Blocking Na/K-ATPase/ROS Amplification in C57Bl6 and ApoE Knockout Mice Fed a Western Diet. Sci Rep 7, 193 (2017).
van der Heijden, R. A. et al. High-fat diet induced obesity primes inflammation in adipose tissue prior to liver in C57BL/6j mice. Aging 7, 256–268 (2015).
Tchkonia, T. et al. Fat tissue, aging, and cellular senescence. Aging Cell 9, 667–684 (2010).
Vidal-Puig, A. et al. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J Clin Invest 97, 2553–2561 (1996).
Matos, L., Gouveia, A. & Almeida, H. Copper ability to induce premature senescence in human fibroblasts. Age (Dordr) 34, 783–794 (2012).
Gire, V., Roux, P., Wynford-Thomas, D., Brondello, J. M. & Dulic, V. DNA damage checkpoint kinase Chk2 triggers replicative senescence. Embo j 23, 2554–2563 (2004).
Chen, Q. & Ames, B. N. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proceedings of the National Academy of Sciences of the United States of America 91, 4130–4134 (1994).
Chen, H., Li, Y. & Tollefsbol, T. O. Cell senescence culturing methods. Methods Mol Biol 1048, 1–10 (2013).
Kumazaki, T., Robetorye, R. S., Robetorye, S. C. & Smith, J. R. Fibronectin expression increases during in vitro cellular senescence: correlation with increased cell area. Experimental cell research 195, 13–19 (1991).
Chen, Q. M. Replicative senescence and oxidant-induced premature senescence. Beyond the control of cell cycle checkpoints. Annals of the New York Academy of Sciences 908, 111–125 (2000).
Kuo, L. J. & Yang, L. X. Gamma-H2AX - a novel biomarker for DNA double-strand breaks. In vivo 22, 305–309 (2008).
Kyrylkova, K., Kyryachenko, S., Leid, M. & Kioussi, C. Detection of apoptosis by TUNEL assay. Methods in molecular biology 887, 41–47 (2012).
Wurstle, M. L., Laussmann, M. A. & Rehm, M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Experimental cell research 318, 1213–1220 (2012).
Jena, N. R. DNA damage by reactive species: Mechanisms, mutation and repair. Journal of biosciences 37, 503–517 (2012).
Hemnani, T. & Parihar, M. S. Reactive oxygen species and oxidative DNA damage. Indian journal of physiology and pharmacology 42, 440–452 (1998).
Bennett, M. R. Reactive oxygen species and death: oxidative DNA damage in atherosclerosis. Circulation research 88, 648–650 (2001).
Campisi, J. Aging, cellular senescence, and cancer. Annual review of physiology 75, 685–705 (2013).
Davalli, P., Mitic, T., Caporali, A., Lauriola, A. & D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxidative medicine and cellular longevity 2016, 3565127 (2016).
Liu, D. & Xu, Y. p53, oxidative stress, and aging. Antioxid Redox Signal 15, 1669–1678 (2011).
Scholzen, T. & Gerdes, J. The Ki-67 protein: from the known and the unknown. Journal of cellular physiology 182, 311–322 (2000).
Bruey, J. M. et al. Circulating Ki-67 index in plasma as a biomarker and prognostic indicator in chronic lymphocytic leukemia. Leukemia research 34, 1320–1324 (2010).
Kim, H., Yoon, S. C., Lee, T. Y. & Jeong, D. Discriminative cytotoxicity assessment based on various cellular damages. Toxicology letters 184, 13–17 (2009).
Loo, A. E. & Halliwell, B. Effects of hydrogen peroxide in a keratinocyte-fibroblast co-culture model of wound healing. Biochemical and biophysical research communications 423, 253–258 (2012).
Quan, C., Cho, M. K., Perry, D. & Quan, T. Age-associated reduction of cell spreading induces mitochondrial DNA common deletion by oxidative stress in human skin dermal fibroblasts: implication for human skin connective tissue aging. Journal of biomedical science 22, 62 (2015).
Marthandan, S. Effects of ebselen and N-acetyl cysteine on replicative aging of primary human fibroblast strains. Immun Ageing 12, 8 (2015).
La Fata, G., Seifert, N., Weber, P. & Mohajeri, M. H. Vitamin E Supplementation Delays Cellular Senescence In Vitro. BioMed research international 2015, 563247 (2015).
Butt, H. et al. Protective role of vitamin E preconditioning of human dermal fibroblasts against thermal stress in vitro. Life sciences 184, 1–9 (2017).
Zeng, J. P. et al. Repeated exposure of mouse dermal fibroblasts at a sub-cytotoxic dose of UVB leads to premature senescence: a robust model of cellular photoaging. J Dermatol Sci 73, 49–56 (2014).
Liu, J., Yan, Y., Nie, Y. & Shapiro J. I. Na/K-ATPase Signaling and Salt Sensitivity: The Role of Oxidative Stress. Antioxidants (Basel) 6, (2017).
Foyouzi, N. et al. Effects of oxidants and antioxidants on proliferation of endometrial stromal cells. Fertil Steril 82(Suppl 3), 1019–1022 (2004).
Alcendor, R. R. et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circulation research 100, 1512–1521 (2007).
Petropoulou, C., Trougakos, I. P., Kolettas, E., Toussaint, O. & Gonos, E. S. Clusterin/apolipoprotein J is a novel biomarker of cellular senescence that does not affect the proliferative capacity of human diploid fibroblasts. FEBS letters 509, 287–297 (2001).
Aravinthan, A. et al. The senescent hepatocyte gene signature in chronic liver disease. Experimental gerontology 60, 37–45 (2014).
Sebastian, C. et al. Telomere shortening and oxidative stress in aged macrophages results in impaired STAT5a phosphorylation. Journal of immunology (Baltimore, Md.: 1950) 183, 2356–2364 (2009).
Jun, J. I. & Lau, L. F. CCN2 induces cellular senescence in fibroblasts. Journal of cell communication and signaling 11, 15–23 (2017).
Rodier, F. et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nature cell biology 11, 973–979 (2009).
Nguyen, A. N. et al. Ouabain activates the Na-K-ATPase signalosome to induce autosomal dominant polycystic kidney disease cell proliferation. American journal of physiology. Renal physiology 301, F897–906 (2011).
Haas, M., Wang, H., Tian, J. & Xie, Z. Src-mediated inter-receptor cross-talk between the Na+/K+ -ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J Biol Chem 277, 18694–18702 (2002).
Soto-Gamez, A. & Demaria, M. Therapeutic interventions for aging: the case of cellular senescence. Drug Discov Today 22, 786–795 (2017).
Kirkland, J. L., Tchkonia, T., Zhu, Y., Niedernhofer, L. J. & Robbins, P. D. The Clinical Potential of Senolytic Drugs. J Am Geriatr Soc 65, 2297–2301 (2017).
Das, A. et al. Impairment of an Endothelial NAD(+)-H2S Signaling Network Is a Reversible Cause of Vascular. Aging. Cell 173, 74–89 e20 (2018).
Kim, I. H. et al. Aging increases the susceptibility of hepatic inflammation, liver fibrosis and aging in response to high-fat diet in mice. Age (Dordrecht, Netherlands) 38, 291–302 (2016).
Kennedy, D. J. et al. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am J Physiol Renal Physiol 294, F450–454 (2008).
Haller, S. T. et al. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. American Journal of Hypertension 25, 690–696 (2012).
Wouters, K. et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 48, 474–486 (2008).
Lazarus, R. C., Buonora, J. E., Jacobowitz, D. M. & Mueller, G. P. Protein carbonylation after traumatic brain injury: cell specificity, regional susceptibility, and gender differences. Free radical biology & medicine 78, 89–100 (2015).
Zheng, J. & Bizzozero, O. A. Accumulation of protein carbonyls within cerebellar astrocytes in murine experimental autoimmune encephalomyelitis. Journal of neuroscience research 88, 3376–3385 (2010).
Shibutani, M., Uneyama, C., Miyazaki, K., Toyoda, K. & Hirose, M. Methacarn Fixation: A Novel Tool for Analysis of Gene Expressions in Paraffin-Embedded Tissue Specimens. Lab Invest 80, 199–208 (0000).
Singh, S. P. et al. PGC-1 alpha regulates HO-1 expression, mitochondrial dynamics and biogenesis: Role of epoxyeicosatrienoic acid. Prostaglandins & other lipid mediators 125, 8–18 (2016).
Lu, H. & Wang, B. SIRT1 exerts neuroprotective effects by attenuating cerebral ischemia/reperfusion-induced injury via targeting p53/microRNA-22. Int J Mol Med 39, 208–216 (2017).
Wang, J. et al. Chlorogenic acid prevents isoproterenol-induced DNA damage in vascular smooth muscle cells. Mol Med Rep 14, 4063–4068 (2016).
Hollingworth, R., Horniblow, R. D., Forrest, C., Stewart, G. S. & Grand, R. J. Localization of Double-Strand Break Repair Proteins to Viral Replication Compartments following Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus. J Virol 91 (2017).
Yan, Y. et al. Ouabain-stimulated trafficking regulation of the Na/K-ATPase and NHE3 in renal proximal tubule cells. Molecular and cellular biochemistry 367, 175–183 (2012).
Zampieri, M. et al. Validation of suitable internal control genes for expression studies in aging. Mech Ageing Dev 131, 89–95 (2010).
Chen, J., Rider, D. A. & Ruan, R. Identification of valid housekeeping genes and antioxidant enzyme gene expression change in the aging rat liver. J Gerontol A Biol Sci Med Sci 61, 20–27 (2006).
van Raalte, H. & Egorov, V. Tactile Imaging Markers to Characterize Female Pelvic Floor Conditions. Open J Obstet Gynecol 5, 505–515 (2015).
Al-Zuhir, N. et al. Selection, thirty day outcome and costs for short stay endovascular aortic aneurysm repair (SEVAR). European journal of vascular and endovascular surgery: the official journal of the European Society for Vascular Surgery 43, 662–665 (2012).
Dial, L., Liu, J. & Shapiro, J. I. Cardiotonic steroids in adaptation to dietary salt intake. Curr Clin Pharmacol 9, 298–309 (2014).
Acknowledgements
This work was supported by National Institutes of Health Grants HL109015 (to J.I.S. and Z.X.), HL071556 and HL105649 (to J.I.S.), HL55601, DK106666-01 (J.L.), and HL34300 (to N.G.A.), by the Brickstreet Foundation (to J.I.S. and N.G.A.) and by the Huntington Foundation, Inc. We acknowledged David Neff from the Molecular and Biological Imaging Center, at Marshall University, for his contribution to the confocal imaging. We also acknowledged Jung Han Kim from Department of Biomedical Sciences, for her contribution in the measurement of oxygen consumption and energy expenditure in the mice.
Author information
Authors and Affiliations
Contributions
Komal Sodhi: Designed and performed the experiments, analyzed data and participated in the writing of the manuscript. Alexandra Nichols, Rebecca L. Klug, Xiaoliang Wang, Krithika Srikanthan, Perrine Goguet-Rubio, Athar Nawab, Rebecca Pratt and Megan N. Lilly: Performed experiments and analyzed data. Amrita Mallick: Performed the experiments and participated in experimental design. Jiang Liu: Performed experiments and analyzed data. Also participated in experimental design. Juan R. Sanabria: Edited the manuscript and participated in data analysis. Zijian Xie and Nader G. Abraham: Participated in the design of experiments and editing of the manuscript. Joseph I. Shapiro: Conceived and designed the overall project as well as individual experiments, analyzed data and served as senior author in the writing of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
Several of the authors (Komal Sodhi, Jiang Liu, Zijian Xie, Nader Abraham and Joseph I. Shapiro) have patents awarded and/or patent applications, which may ultimately constitute a competing financial interest. The authors do not have any non-financial competing interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been retracted. Please see the retraction notice for more detail:https://doi.org/10.1038/s41598-022-13456-y
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sodhi, K., Nichols, A., Mallick, A. et al. RETRACTED ARTICLE: The Na/K-ATPase Oxidant Amplification Loop Regulates Aging. Sci Rep 8, 9721 (2018). https://doi.org/10.1038/s41598-018-26768-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-26768-9
This article is cited by
-
The Na/K-ATPase Signaling and SGLT2 Inhibitor-Mediated Cardiorenal Protection: A Crossed Road?
The Journal of Membrane Biology (2021)
-
RETRACTED ARTICLE: The Adipocyte Na/K-ATPase Oxidant Amplification Loop is the Central Regulator of Western Diet-Induced Obesity and Associated Comorbidities
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
