
Abstract
A prospective analysis investigating the associations between pathogenic copy number variations (pCNVs) and ultrasound soft markers (USMs) in fetuses and evaluating the clinical value of copy number variation sequencing (CNV-seq) in such pregnancy studies was carried out. 3,398 unrelated Chinese women with singleton pregnancies and undergone amniocentesis at 18–36 weeks of gestation for fetal CNV-seq were included. According to the prenatal fetal ultrasound screening results, the samples were divided into 3 groups: normal ultrasound (n = 2616), solitary USM (n = 663), and two or more USMs (n = 119). CNV-seq was performed successfully using all samples. The prevalence of pCNVs in fetuses with normal ultrasound and USMs was 3.03% (79/2616) and 2.94% (23/782), respectively. The risk of segmental aneuploidies was significantly higher in the two or more USMs group (5/119, 4.20%) than in the normal ultrasound (27/2616, 1.04%) or solitary USM (9/663, 1.36%) groups (p = 0.002 and p = 0.031, respectively). Assuming that the resolution of karyotyping is ~5 Mb, a cytogenetic analysis would miss 33 of 102 (32.35%) pCNVs in these samples. Our results suggest an association between pCNVs and fetal USMs; multiple USMs indicate an increased risk of fetal segmental aneuploidies. In prenatal diagnostic testing, CNV-Seq identified additional, clinically significant cytogenetic information.
Similar content being viewed by others

Introduction
Ultrasound has become a crucial tool in the field of fetal medicine. In particular, first and second trimester scans have become a routine part of antenatal care to detect congenital anomalies that are solitary or part of an underlying chromosomal anomaly. Minor sonographic features of fetal aneuploidy, most commonly Down Syndrome (DS), were first reported in the 1980s1. As ultrasound technology has advanced, the detection of these soft markers has become easier. A large number of other ultrasound findings have been reported to be associated with common trisomy 21, 18, and 13, including nuchal fold thickening, ventriculomegaly, short femur or humerus, hypoplastic nose, echogenic bowel, pyelectasis, sandal gap toes, choroid plexus cysts, enlarged cisterna magna, single umbilical artery, and echogenic intracardiac foci2,3. Some soft markers have no significance beyond the association with aneuploidy, while some have potential implications that require evaluation and follow-up4,5. However, Some scholars have recommended that, when an abnormality/marker is detected at routine ultrasound examination, It is best to base counseling on an individual estimated risk for a chromosomal defect, rather than the arbitrary advice that invasive testing is recommended because the risk is high6. Therefore, the increased detection of ultrasound soft markers (USMs) can lead to confusion regarding the appropriate parental counseling and fetal management. Even if the pregnant woman has undergone amniocentesis or chorionic villus sampling (CVS) because of the presence of a USM in the fetus, the most common follow-up method reported in the literatures was karyotyping, which does not routinely detect small genomic deletions and duplications.
Copy number variations (CNVs) indicate regions of the DNA sequence with gains or losses and occur at high frequency in the human genome; these CNVs can affect the whole or segmental genome7. CNVs can be either benign or pathogenic, depending on their location and genetic content. Pathogenic CNVs (pCNVs) are increasingly associated with complicated diseases, especially with some structural congenital anomalies8. A recent study suggests an association between pCNVs and fetal isolated mild ventriculomegaly; pCNVs may also be involved in the pathological process of fetal isolated mild ventriculomegaly and postnatal neurodevelopmental disorders9. However, limited data exist on whether the presence of other USMs indicates an increased risk of pCNVs, except for common trisomy 21, 18, and 13. The potential association between pCNVs and fetal USMs is unclear, especially as this association relates to segmental aneuploidies.
The fetal sonographic anatomical screening is usually performed at 22–25 weeks of gestation in China, because many cardiac defects are not detected until relatively late in the second trimester of pregnancy10. Therefore, most USMs are detected at 22–25 weeks of gestation. If these pregnant women choose to diagnose fetal chromosomal abnormalities through prenatal diagnosis, conventional cytogenetic analysis most commonly involves cell culture from the amniotic fluid, but fetal fibroblasts obtained after 24 weeks of gestation take longer to grow (often longer than 2 weeks) with a higher culture failure rate than those sampled earlier in gestation11. Fetal blood sampling by puncture of the umbilical cord vein provides an alternative, quicker method for karyotyping in late pregnancy, but it is technically more difficult and has a higher miscarriage rate. Many prenatal services have developed multiplex fluorescence in situ hybridization (FISH), quantitative fluorescence polymerase chain reaction (QF-PCR), bacterial artificial chromosome on beads (BOBs), or multiplex ligation-dependent probe amplification (MLPA) for amniocenteses collected after 24 weeks, but these approaches are limited by the number of target chromosomes tested per assay.
Array comparative genomic hybridization (aCGH), first proposed in 199712, has served as a robust and effective approach to screen for CNVs13. However, aCGH is expensive and has limited resolution and accuracy. Today, rapidly developing next generation sequencing (NGS) technologies provide a sensitive and accurate alternative approach to assess genomic variations. The quality, speed, and affordability give NGS a significant advantage over microarrays14,15,16. Thus far, there is little data reporting the use of NGS copy number variation sequencing (CNV-seq) for prenatal karyotyping in fetuses with USMs. In this prospective study, we analyze the frequency of pCNVs in fetuses with USMs and evaluate the application of CNV-seq as a tool for the identification of pCNVs.
Results
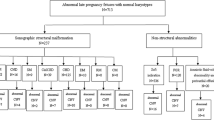
Prospectively, we studied 3,398 unrelated Chinese women with a singleton pregnancy who were referred for an amniocentesis procedure to investigate the presence of a potential chromosome abnormality in the fetus. Based on the prenatal fetal ultrasound screening results, the samples were divided into 3 groups: normal ultrasound (n = 2616), solitary USM (n = 663), or two or more USMs (n = 119) (Table 1). CNV-seq was performed for all samples successfully. Overall, CNV-Seq revealed that 3,296 samples were euploid (97.00%) and 102 samples contained pCNVs (3.00%). Of these 102 cases with pCNVs, 33 (32.35%) had submicroscopic CNVs (smaller than 5 Mb).
The mean gestational age and maternal age at the time of amniocentesis for the group with normal ultrasound results (n = 2,616) were 21.8 weeks (range 18.2–35.6) and 32.0 years (range 15.0–46.0), respectively. In this group, a total of 79 pCNVs (3.03%) were detected; of these, 27 (34.18%) had segmental aneuploidies. Additionally, 782 fetuses presenting with USMs (either solitary USM or multiples USMs) were included in this study. The mean gestational age and maternal age at the time of amniocentesis were 26.3 weeks (range 18.5–36.0) and 28.2 years (range 17.0–45.0), respectively. The most common USM was echogenic focus in the heart (486 samples, 62.15%), followed by mild ventriculomegaly (67 samples, 8.57%) and mild pyelectasis (62 samples, 7.93%). A total of 23 pCNVs (2.94%) were detected from these 782 fetuses. In samples from the 663 fetuses with a solitary USM, 17 (2.56%) featured pCNVs. Of these, 9 (52.94%) were segmental aneuploidies. Out of the 119 fetuses with two or more USMs, 6 pCNVs (5.04%) were detected, and 5 were segmental aneuploidies.
The risk of segmental aneuploidies was significantly increased in the group with two or more USMs (5/119, 4.20%) when compared with the group with normal ultrasound (27/2616, 1.04%) or a solitary USM (9/663, 1.36%) (p = 0.002 and p = 0.031, respectively). Out of the 782 fetuses with USMs, 9 whole chromosome aneuploidies were found in 9 cases (1.15%); 4 were DS, 2 were T18, and the remaining 3 featured different types of sex chromosome aneuploidies. These whole chromosome aneuploidies were all confirmed by QF-PCR or FISH (Table 2). Also, there were 14 cases (1.79%) with segmental aneuploidies; of these, 10 cases (10/14, 71.43%) featured microdeletions or microduplications associated with known chromosome disease syndromes. All segmental aneuploidies were confirmed by karyotyping or aCGH (Table 3). Most patients (8/14, 57.14%) with a fetus with segmental aneuploidies consented to parental gDNA studies to complete a full trio analysis. It was found that 5 of the segmental aneuploidies were formed de novo (5/8, 62.5%) and 3 were inherited (3/8, 37.5%).
Discussion
Chromosomal abnormalities are a serious threat to human health; some fetuses with chromosomal abnormalities have obvious structural abnormalities by ultrasound, but some do not. USMs are often found in fetuses with whole chromosome aneuploidy (mostly in DS) and congenital structural anomalies. However, USMs can also be found as normal variants in a substantial proportion of euploid fetuses without structural anomalies. USMs are detected during fetal sonographic anatomical screening in approximately 10% of normal fetuses in the USA17. Ginsberg showed that there is a high recurrence rate of solitary USMs, which supports the hypothesis that genetic factors form the basis for the reappearance of USMs18. Therefore, there were different incidences of USMs among fetuses of different races.
In our study of a population of 3,398 singleton pregnancies, USMs were detected in 23.01% of the fetuses. CNV-Seq detected 9 whole chromosome aneuploidies from the 782 fetuses with USMs, and DS was the most common abnormality. These results coincide with those reported previously in the literatures1,19. The incidence of pCNVs in fetuses with USMs was 2.94% (23/782), which is similar to that in fetuses with a normal ultrasound (79/2616, 3.03%). The group with normal ultrasound results featured patients with advanced maternal age or a high risk for chromosomal abnormality based on maternal serum screening; these are high risk factors for whole chromosome aneuploidies20. The estimated prevalence of chromosomal abnormalities is 0.1–0.65% for newborns21,22. Our findings show that the presence of USMs is associated with an increased risk of pCNVs compared to the risk in the general pregnant population.
In the 782 samples with USMs in our study, whole chromosome aneuploidies did not increase as the number of USMs increased. The frequencies of segmental aneuploidies in the normal ultrasound, solitary USM, and two or more USMs groups were 1.04%, 1.36%, and 4.20%, respectively. The risk of segmental aneuploidies in the two or more USMs group was significantly increased compared to the risk in the other groups. Accordingly, our results suggest that there is an association between segmental aneuploidies and multiple USMs, which may be a useful prenatal clinical indicator for segmental aneuploidies.
Based on the assumption that the resolution of karyotyping is on the order of 5 Mb, a cytogenetic analysis of these 3,398 samples would have detected all 61 whole chromosome aneuploidies, but missed 33 of the 41 pathogenic segmental aneuploidies, giving a detection rate of 2.03% (69/3398). In contrast, the detection rate using CNV-Seq was 3.00% (102/3398), potentially increasing the detection of pCNVs by 0.97% when compared to karyotyping. Submicroscopic CNVs are an important cause of developmental delay, intellectual disability, and autism spectrum disorders in children23. In the context of prenatal diagnostic testing, high resolution detection of chromosome abnormalities identified additional, clinically significant cytogenetic information that was missed with karyotyping24.
At the time of this study, 4 of the 23 fetuses with pCNVs detected with USMs had not been terminated; 3 fetuses have been born and examined by a pediatrician to reveal no obvious features of a disease syndrome. However, because the children are very young and the clinical phenotype may not have appeared yet, a follow-up is necessary for these children. In the 14 fetuses with segmental aneuploidies, obvious structural abnormalities were detected in 3 fetuses by subsequent ultrasound: 1 case (P3) featured bilateral renal enlargement with echo enhancement, 1 case featured bilateral small kidneys (P4), and 1 case (P20) featured a ventricular septal defect, pulmonary stenosis, and polyhydramnios. The amniocytes from P20 were 47, XX, t(1; 15) (q32.2; q13), +der (15) t(1; 15) (q32.2; q13)mat, whereas the karyotype of the mother was 46, XX, t(1; 15) (q32.2; q13), and the father showed the normal karyotype 46, XY. The couple already had a 12-year old daughter with a normal phenotype who had not yet undergone a chromosome analysis. The CNV-seq profiles and partial karyograms of the fetus are shown in Fig. 1. Additionally, a fetus (P8) with dup3q26.33q29 (15.28 Mb) and del10q26.3 (2.54 Mb) because of a maternal balanced reciprocal translocation was detected. Finally, P5 featured a 1q21.1 recurrent microdeletion, which was inherited from the mother, whose only phenotype was irritability.

The CNV-seq profiles and partial karyograms of the fetus. (A,B) CNV-seq profiles for dup1q32.2q44 and dup15q11.1q13.2 (arrows), data is plotted as copy number (Y-axis) versus 20 kb chromosomal read bins (X-axis). The mean copy number along the length of each chromosome is indicated by the blue line. (C,D) The amniocytes of P20 showed partial karyotype and ideogram with an additional derivative chromosome der (15) t(1; 15) (q32.2; q13).
USMs of aneuploidy were originally proposed to assess the risk for DS in women of advanced maternal age. Current ultrasound and serum screening protocols have far better sensitivity and specificity than age alone25, and screening by the analysis of cell-free DNA in the maternal blood in singleton pregnancies can detect >99% of fetuses with DS, 98% with T18, and 99% with T1326. When the aneuploidy risk is no longer a concern, follow-up for such patients should include consideration of other non-aneuploidy-related associations, such as chromosomal structural abnormalities and congenital structural anomalies.
In conclusion, our results suggest that there is an association between pCNVs and fetus USMs; multiple USMs are associated with an increased risk of fetal segmental aneuploidies. Identifying specific genomic alterations may provide insight into the pathogenetic mechanism and aid in better diagnosis and prognosis for adverse outcomes from fetal USMs. The accuracy of pCNV detection by CNV-Seq in 102 patients was also assessed using a range of other gold-standard molecular techniques. All confirmatory results from the validation tests were consistent with the original CNV-Seq diagnoses, indicating a 100% concordance rate. In the context of prenatal diagnostic testing, CNV-Seq identified additional, clinically significant cytogenetic information that was missed with karyotyping.
Materials and Methods
Biological Samples
This study involved pregnant women who underwent amniocentesis at 18–36 weeks of gestation for fetal CNV-seq at the West China Second University Hospital of Sichuan University between February and September 2017. None of the fetuses showed any other structural abnormalities by ultrasonogram before amniocentesis. Samples were categorized into 3 different fetal risk groups: fetuses with normal ultrasound, solitary USM, or two or more USMs. The fetal group with normal ultrasound consisted of patients with advanced maternal age (AMA, over 35 years at the expected date of childbirth) and high risk for DS or trisomy 18 (T18) based on maternal serum screening. USMs included thickened nuchal fold, echogenic bowel, mild ventriculomegaly (range 10–15 mm)27, echogenic focus in the heart, choroid plexus cyst, enlarged cisterna magna, mild pyelectasis (dilatation of the renal pelvis ≥4 mm in the 2nd trimester and ≥7 mm thereafter), absence or dysplasia of nasal bone, short long bones, tricuspid regurgitation, persistent left superior vena cava, and umbilical cord abnormality (single umbilical artery or dilatation of the umbilical vein). The diagnosis of USMs was confirmed by a second experienced ultrasonographer. As a quality control measure for the amniocyte DNA, maternal DNA contamination from the procedure was assessed by QF-PCR using a set of 15 highly polymorphic short tandem repeat (STR) markers in all samples. If the sample was deemed to have maternal DNA contamination, the amniocyte DNA sample was considered unacceptable for a reliable chromosome test result. The other amniotic fluid samples from the patient were used for cell culture following the standard protocols28, and the CNVs were analyzed from the genomic DNA (gDNA) isolated from the cultured amniotic fluid cells. The study was approved by the Institutional Ethics Committee of Sichuan University, and test results were used for research with the informed consent of the parents. We confirm that all methods were performed in accordance with the relevant guidelines and regulations.
Copy Number Variation Sequencing (CNV-seq)
RNA-free high-molecular-weight DNA was isolated from amniotic fluid using the DNeasy Blood and Tissue Kit (Qiagen GmbH, Hilden, Germany). The quality and concentration of gDNA from the samples was assessed using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA). gDNA (50 ng) was fragmented, and DNA libraries were constructed by end filling, adapter ligation, and PCR amplification. DNA libraries were subjected to massively parallel sequencing using the NextSeq CN500 platform (Illumina) to generate 5 million raw sequencing reads with 36-bp genomic DNA sequences. Using the hg19 genomic sequence as a reference, a total of 2.8–3.2 million reads were uniquely and precisely mapped using the Burrows Wheelers algorithm29. Mapped reads were allocated progressively to 20 kb bin sizes from the p to q arms of the 24 chromosomes. Counts in each bin were then compared between all test samples run in the same flow cell to evaluate copy number changes using previously described algorithms30,31. Chromosome profiles were finally plotted as copy number (Y-axis) versus 20 kb count window (X-axis). A blue line was used to indicate the mean copy number across each chromosome to identify the nature and map position of any deleted or duplicated regions. Identified and mapped CNVs were interrogated using publicly available databases, including DECIPHER (https://decipher.sanger.ac.uk/), DGV (http://dgv.tcag.ca/), 1000 genomes (http://www.internationalgenome.org/), OMIM (http://omim.org/), and PubMed (https://www.ncbi.nlm.nih.gov/pubmed). CNVs were classified into three categories: benign CNVs, variants of uncertain significance (VOUS), and pCNVs. CNVs coinciding with known polymorphic CNVs were interpreted as benign. CNVs were defined as pathogenic if they were identified in regions of known clinical relevance or in genes associated with known disease phenotypes. CNVs that did not fit any of the above criteria were categorized as VOUS. In this study, we only focused on pCNVs. Additionally, we examined parental gDNA (if available) to determine if the pCNVs identified were formed de novo or inherited.
Confirmatory testing of pCNVs
QF-PCR for chromosomes 13, 18, 21, X, and Y was performed for all samples. The STR markers used were D21S1435, D21S1411, and D21S11 for chromosome 21; D18S1002, D18S391, D18S535, and D18S386 for chromosome 18; D13S628, D13S742, D13S634, and D13S305 for chromosome 13; and DXS981, DXS6809, DXS22, and AMXY for the X and Y chromosomes. PCR was performed using a Bio-Rad PTC-200 PCR system (Bio-Rad, Mexico City, Mexico). The PCR products were analyzed using an ABI 3500 automated sequencer (Applied Biosystems, Tokyo, Japan). Sizing and labeling alleles were collected with Gene Mapper software (Applied Biosystems, Foster City, CA, USA). Allele dosage ratios between 0.8 and 1.4 were defined as normal, while an allele ratio >1.8, <0.65, or the presence of three alleles of equal area indicated trisomy. A single peak was described as uninformative32. When all markers for a chromosome were uninformative or the QF-PCR failed to have a result, enumeration was done by FISH (GP Medical, Beijing, China). Amniocytes were assessed for aneuploidy according to the manufacturer’s protocol (http://www.gpmedical.com.cn) as previously described33. Amniotic fluid samples were used for cell culture following the standard protocols28. Cells were cultivated with AmnioMAX-II (Gibco, Carlsbad, Calif., USA) medium. Chromosome preparations were G-banded using trypsin-Giemsa for karyotyping. A minimum of ten metaphases at the 400–500 band resolution level were routinely analyzed for each specimen. Chromosome karyotype map scanning and acquisition were done using an automatic metaphase chromosome analysis system (MetaSystems, Göttingen, Germany). Karyotypes were analyzed by two qualified senior laboratory technicians. aCGH was performed using the CGX v2 Oligo aCGH Kit (manufactured by Agilent Technologies, Santa Clara, CA, USA; Perkin Elmer, Turku, Finland). The microarray was scanned using the Agilent SureScan Microarray Scanner (Agilent, Santa Clara, CA, USA). Data were extracted using the Agilent CytoGenomics software (Agilent, Santa Clara, CA, USA) and analyzed using the Genoglyphix Analysis software (Perkin Elmer, Waltham, MA, USA).
References
Benacerraf, B. R., Barss, V. A. & Laboda, L. A. A sonographic sign for the detection in the second trimester of the fetus with Down’s syndrome. Am J Obstet Gynecol 151, 1078–1079 (1985).
Nyberg, D. A. & Souter, V. L. J. Sonographic markers of fetal trisomies: second trimester. Ultrasound Med 20, 655–674 (2001).
Benacerraf, B. R. et al. Fetal pyelectasis: a possible association with Down syndrome. Obstet Gynecol 76, 58–60 (1990).
Wollenberg, A., Neuhaus, T. J., Willi, U. V. & Wisser, J. Outcome of fetal renal pelvic dilatation diagnosed during the third trimester. Ultrasound Obstet Gynecol 25, 483–488 (2005).
Norton, M. E. Follow-up of sonographically detected soft markers for fetal aneuploidy. Semin Perinatol 37, 365–369 (2013).
Nicolaides, K. H. & Wegrzyn, P. Sonographic features of chromosomal defects in the second trimester of pregnancy. Ginekol Pol 76, 528–535 (2005).
Redon, R. et al. Global variation in copy number in the human genome. Nature 444, 444–454 (2006).
Mccarroll, S. A. & Altshuler, D. M. Copy-number variation and association studies of human diseases. Nature Genetics 39, 37–42 (2007).
Hu, P. et al. Copy Number Variations with Isolated Fetal Ventriculomegaly. Curr Mol Med 17, 133–139 (2017).
Yagel, S. et al. Congenital heart defects: natural course and in utero development. Circulation 96, 550–555 (1997).
Lam, Y. H., Tang, M. H., Sin, S. Y. & Ghosh, A. Clinical significance of amniotic-fluid-cell culture failure. Prenat Diagn 18, 343–347 (1998).
Solinas-Toldo, S. et al. Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer 20, 399–407 (1997).
Feuk, L., Carson, A. R. & Scherer, S. W. Structural variation in the human genome. Nat Rev Genet 7, 85–97 (2006).
Hurd, P. J. & Nelson, C. J. Advantages of next-generation sequencing versus the microarray in epigenetic research. Brief Funct Genomic Proteomic 8, 174–183 (2009).
Su, Z. et al. Comparing next-generation sequencing and microarray technologies in a toxicological study of the effects of aristolochic acid on rat kidneys. Chem Res Toxicol 24, 1486–1493 (2011).
Zhu, X. et al. Identification of copy number variations associated with congenital heart disease by chromosomal microarray analysis and next-generation sequencing. Prenat Diagn 36, 321–327 (2016).
Nyberg, D. A., Luthy, D. A., Resta, R. G., Nyberg, B. C. & Williams, M. A. Age-adjusted ultrasound risk assessment for fetal Down’s syndrome during the second trimester: description of the method and analysis of 142 cases. Ultrasound Obstet Gynecol 12, 8–14 (1998).
Ginsberg, Y., Khatib, N., Weiner, Z., Beloosesky, R. & Bronshtein, M. The recurrence of sonographic ‘soft markers’: ominous sign or ‘just’ genetics? Prenat Diagn 37, 469–472 (2017).
Agathokleous, M., Chaveeva, P., Poon, L. C., Kosinski, P. & Nicolaides, K. H. Meta-analysis of second-trimester markers for trisomy 21. Ultrasound Obstet Gynecol 41, 247–261 (2013).
Duncan, F. E. et al. Chromosome cohesion decreases in human eggs with advanced maternal age. Aging Cell 11, 1121–1124 (2012).
American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 88, December 2007. Invasive prenatal testing for aneuploidy. Obstet Gynecol 110, 1459–1467 (2007).
Hernandez-Herrera, R. J. et al. Chromosomal abnormalities in patients from Obstetrics and Gynaecology hospital. Rev Med Inst Mex Seguro Soc 52, 90–93 (2014).
Cooper, G. M. et al. A copy number variation morbidity map of developmental delay. Nat Genet 43, 838–846 (2011).
Wapner, R. J. et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 367, 2175–2184 (2012).
Russo, M. L. & Blakemore, K. J. A historical and practical review of first trimester aneuploidy screening. Semin Fetal Neonatal Med 19, 183–187 (2014).
Gil, M. M., Accurti, V., Santacruz, B., Plana, M. N. & Nicolaides, K. H. Analysis of cell-free DNA in maternal blood in screening for aneuploidies: updated meta-analysis. Ultrasound Obstet Gynecol 50, 302–314 (2017).
Senat, M. V., Bernard, J. P., Schwärzler, P., Britten, J. & Ville, Y. Prenatal diagnosis and follow-up of 14 cases of unilateral ventriculomegaly. Ultrasound Obstet Gynecol 14, 327–332 (1999).
Steele, M. W. & Breg, W. R. Jr. Chromosome analysis of human amniotic-fluid cells. Lancet 1, 383–385 (1966).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Liang, D. et al. Copy number variation sequencing for comprehensive diagnosis of chromosome disease syndromes. J Mol Diagn 16, 519–526 (2014).
Wang, Y. et al. Maternal mosaicism is a significant contributor to discordant sex chromosomal aneuploidies associated with noninvasive prenatal testing. Clin Chem 60, 251–259 (2014).
Mann, K. et al. Development and implementation of a new rapid aneuploidy diagnostic service within the UK National Health Service and implications for the future of prenatal diagnosis. Lancet 358, 1057–1061 (2001).
Committee on the Application of Fluorescence In Situ Hybridization Technique in Prenatal Diagnosis. Common opinion on the application of fluorescence in situ hybridization technique in prenatal diagnosis. Zhonghua Fu Chan Ke Za Zhi 51, 241–244 (2016).
Acknowledgements
The authors thank the patients and family members who participated in this research program. This study was jointly funded by grants from the Program of Science and Technology Department of Sichuan Province (No. 2016FZ0116), the Research Project of Health and Family Planning Commission of Sichuan Province (No. 16ZD018) and the Applied Basic Research Program of Science and Technology Department of Sichuan Province (No. 2017JY0272).
Author information
Authors and Affiliations
Contributions
J.W. and H.L. designed the concept and the experiment method of the research. J.W., X.C., Q.Z. and H.L. conducted the most of collection for the samples. J.W., L.C., C.Z. and L.W. collected, analyzed and interpreted the data. J.W. wrote the main paper. H.L. supervised the project, had given valuable advices on the proceeding of this work and revised the manuscript. H.X., Y.X., D.Y., Y.Z., F.T., Y.Y., H.Z. and Z.L. contributed to the experiments. All authors discussed the results and commented on the manuscript at all stages.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Chen, L., Zhou, C. et al. Identification of copy number variations among fetuses with ultrasound soft markers using next-generation sequencing. Sci Rep 8, 8134 (2018). https://doi.org/10.1038/s41598-018-26555-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-26555-6
This article is cited by
-
Experience of copy number variation sequencing applied in spontaneous abortion
BMC Medical Genomics (2024)
-
Identification of copy number variations among fetuses with isolated ultrasound soft markers in pregnant women not of advanced maternal age
Orphanet Journal of Rare Diseases (2024)
-
Prenatal diagnosis of ultrasound soft markers in a single medical center of mainland China
Molecular Cytogenetics (2023)
-
Comparison of the combined use of CNV-seq and karyotyping or QF-PCR in prenatal diagnosis: a retrospective study
Scientific Reports (2023)
-
Investigation on combined copy number variation sequencing and cytogenetic karyotyping for prenatal diagnosis
BMC Pregnancy and Childbirth (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
