
Abstract
Chronic rhinosinusitis with nasal polyp (CRSwNP) patients are often characterized by asthma comorbidity and a type-2 inflammation of the sinonasal mucosa. The mucosal microbiota has been suggested to be implicated in the persistence of inflammation, but associations have not been well defined. To compare the bacterial communities of healthy subjects with CRSwNP patients, we collected nasal swabs from 17 healthy subjects, 21 CRSwNP patients without asthma (CRSwNP−A), and 20 CRSwNP patients with co-morbid asthma (CRSwNP+A). We analysed the microbiota using high-throughput sequencing of the bacterial 16S rRNA. Bacterial communities were different between the three groups. Haemophilus influenzae was significantly enriched in CRSwNP patients, Propionibacterium acnes in the healthy group; Staphylococcus aureus was abundant in the CRSwNP−A group, even though present in 57% of patients. Escherichia coli was found in high amounts in CRSwNP+A patients. Nasal tissues of CRSwNP+A patients expressed significantly higher concentrations of IgE, SE-IgE, and IL-5 compared to those of CRSwNP−A patients. Co-cultivation demonstrated that P. acnes growth was inhibited by H. influenzae, E. coli and S. aureus. The nasal microbiota of healthy subjects are different from those of CRSwNP−A and CRSwNP+A patients. However, the most abundant species in healthy status could not inhibit those in CRSwNP disease.
Similar content being viewed by others


Introduction
Chronic rhinosinusitis with nasal polyps (CRSwNP) is defined as a subgroup of chronic rhinosinusitis (CRS) that is characterized by the presence of fleshy swellings (nasal polyps) that develop in the lining of the nose and paranasal sinuses1. Nasal polyps (NP) are believed to arise in the nasal mucosa because of chronic inflammation. In central Europe, CRSwNP is mostly characterized by a moderate to severe T helper type 2 (Th2)-mediated inflammation with hypereosinophilia and increased IgE concentrations. CRSwNP is difficult to treat and recurrences are frequent, despite medical treatment and surgical interventions. In addition, patients suffering from CRSwNP often have comorbid diseases, such as asthma and aspirin intolerance2,3.
The definitive mechanisms underlying the pathogenesis of CRS remain poorly elucidated4. Microbial involvement have been considered to be one of the mechanisms contributing to the inflammation in case of CRS5,6. Whereas in some reports the characteristics of sinus microbiota in CRS were similar to those of control groups7,8,9, other analyses, making use of CRS phenotypes or endotypes, demonstrated different compositions of resident sinus bacterial communities6,10. Our group has previously demonstrated that Staphylococcus aureus is able to drive Th2 type inflammation and that it is associated with Th2-biased CRSwNP5,6,11. We have also reported that the expression of IL-5 and of IgE against S. aureus superantigens (SE-IgE) within polyp tissue is associated with comorbid asthma12,13. Furthermore, because the levels of IL-5 and SE-IgE were significantly increased in recurrent versus non-recurrent CRSwNP14 and because several studies suggest that alterations in the airway microbiota may be associated with inflammatory processes6,7,10,15,16,17. We reasoned that a study with well-defined subgroups of CRS patients could possibly identify different microbiota, which might be related to specific pathological or immunological characteristics of the inflammation.
Here, we hypothesized that heterogeneousness of CRSwNP with regard to asthma comorbidity could be associated with the presence of compositionally distinct sinus microbiota which affect host immune responses. To validate this hypothesis, we collected nasal swabs to obtain a suitable biological sample from the middle meatus, the common location of nasal polyps18, of CRSwNP patients without co-morbid asthma and compared their sinus microbiota to that of patients with CRSwNP with co-morbid asthma and to that of healthy control subjects, using 16S ribosomal RNA-gene (16S rRNA gene) high throughput sequencing. Furthermore, we studied the in vitro inhibitory and/or stimulatory effects of isolated strains of different species on each other, specifically to understand to what extent the species that are most frequently present in controls may protect the mucosa against germs most frequent in patients with CRSwNP.
Materials and Methods
Study Design and Population
We evaluated nasal microbiota from non-asthmatic CRSwNP patients, CRSwNP patients with co-morbid asthma, and healthy control subjects. Diagnosis of CRSwNP was made according to the European Position Paper on Rhinosinusitis and Nasal Polyps19. Diagnosis of asthma was confirmed by a pulmonologist. The atopic status was evaluated by skin prick tests to common inhalant allergens. The control group consisted of healthy volunteers and patients who were free from rhinosinusitis, asthma, and atopy. Patients less than 18 years of age, subjects with cystic fibrosis, known or suspected immunodeficiency or autoimmune disease, and/or suspected of the use of systemic antibiotics or oral steroids in the last 3 months before sample collection, were excluded from this study.
Ethics statement
This study was approved by the ethics committee of the University of Ghent, Belgium and assigned number B670201422215. All participants provided written informed consent prior to their participation in the project. All experiments were performed in accordance with relevant guidelines and regulations.
Sample Collection
Specimens were collected at the Ghent University Hospital, Belgium. Swab specimens for DNA extraction were obtained with eSwab (COPAN, Brescia, Italy). Swabs were endoscopically guided to the middle meatus region and rotated at least 3 times. In addition, nasal tissue samples were obtained from the patients during endonasal sinus surgery. All samples were immediately transported to the laboratory and snap frozen in liquid nitrogen and stored at −80 °C until further analysis.
Measurement of Cytokines in Nasal Tissue Samples
Snap-frozen tissue specimens were weighed and suspended in a ratio 0.1 g of tissue per 1 mL of 0.9% NaCl solution with a complete protease inhibitor cocktail (Roche, Mannheim, Germany). To prepare soluble protein fractions, frozen tissues were mechanically pulverized using a Tissue Lyser LT (Qiagen, Hilden, Germany) at 50 oscillations per second for two minutes in prechilled Eppendorf tubes. Homogenized tissues were centrifuged at 1,800 × g for ten minutes at 4 °C, and the supernatants were collected. Total IgE, eosinophil cationic protein (ECP) and specific IgE to staphylococcal enterotoxins (SE-IgE) were measured using the UniCAP system (Thermo Fisher Scientific, Phadia, Uppsala, Sweden). Tumor necrosis factor-alpha (TNFα), interleukin (IL)-5 and IL-17 were quantified using the Bio-Plex 200 System (Bio-Rad, Temse, Belgium).
DNA Extraction and Bioinformatic Analysis
DNA was extracted from the whole swab using the FastDNA Spin kit (MP Biomedicals, Solon, Ohio) according to the manufacturer’s instructions. Total DNA concentration was measured with a NanoDrop ND-1000 spectrophotometer (Isogen Life Sciences, IJsselstein, The Netherlands). The quality of the extracted DNA was evaluated on a 1% agarose gel. Libraries were prepared as previously described20, using the primers 27F and 338R20 for the V1-V2 region of the 16S rRNA gene. Libraries were sequenced on a MiSeq (Illumina, Hayward, California). Each sequence was identified using the SILVA database21. Definition of operational taxonomic units (OTUs) and data-set quality filter was performed as previously described20. After quality filtering, the dataset was then filtered to consider only those OTUs sequences that were present in at least one sample at a relative abundance of >0.1% or that were present in all samples at a relative abundance of >0.001%. All samples were randomly re-sampled to equal the smallest read size of 6,682 reads, using the PhyloSeq package22 from the free software R package for statistical computing and graphics23. All the reads were grouped into 942 sequences. All sequences were assigned a taxonomic affiliation (phylotype) based on the naive Bayesian classification (RDP classifier)24. A genus name was assigned to a sequences when only 16S rRNA gene fragments of previously described isolates belonging to that genus and of 16S rRNA gene fragments originating from uncultured representatives of that genus showed ≤2 mismatches. Species assignments were performed using the Basic Local Alignment Search Tool (BLAST). Assignation of a name demanded at least 99% sequence identity over 95% of sequence length25.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism version 6.00 for Mac OS X (GraphPad Software, La Jolla, CA: www.graphpad.com). The categorical outcomes (presence/absence of condition) were expressed as frequencies or percentages and were analyzed using the Chi-square test. The interval data (age, level of cytokines) were tested for distribution using the Shapiro-Wilk normality test. Assessment of the significance of intergroup correlation was performed using the parametric test for Gaussian distributed data and a non-parametric method for non-normally distributed data. The ecological profiles were analyzed using the PAST3 program26. All Principal Coordinate Analyses (PCoA) were based on a Bray-Curtis similarity index, which operated at the phylotype level. To test for community compositional differences, permutation multivariate analysis of variance (PERMANOVA) was employed. Similarity Percentage (SIMPER) analysis was used to determine the contribution of each species to the observed dissimilarity between samples and to identify the species that are most important in creating the observed pattern. In each group, the 20 bacterial species with the highest relative abundance were selected. This resulted in 22 different bacterial species for further analyses. The difference of relative abundance was calculated using a nonparametric Kruskal-Wallis test and corrections of significance for between-group comparisons were calculated using Dunn’s test. The Spearman’s rank correlation was employed to assess the statistical correlation between cytokine and bacterial relative abundance. The statistical significance level was determined as p < 0.05.
Bacterial Cultivation and Interaction Assays
An agar diffusion test (Kirby–Bauer method) was employed to determine in-vitro interaction between bacterial strains. The bacterial strains were selected based on metagenomic and clinical data. In a separate group of patients (10 healthy control subjects, 10 CRSwNP−A patients, and 10 CRSwNP+A patients) we found that the majority of S. aureus strains isolated from CRSwNP patients expressed accessory gene regulator (agr) group I and agr group II. Reference strains expressing agr group I and II were therefore used in this assay. Staphylococcus aureus (gfp RN6390 and ATCC 29213), Escherichia coli (O157:H7 strain), Haemophilus influenzae (ATCC 49144), Corynebacterium pseudodiphtheriticum (CIP 103420T), and Propionibacterium acnes (ATCC 6919) were selected for the experiment. Chocolate agar plate inoculation and incubation at 37 °C with 5% CO2 was determined to be the culture condition that enabled proficient growth of all bacterial strains. Bacterial colonies were harvested, resuspended in 0.9% NaCl to a turbidity that was adjusted to a 0.5 McFarland standard. To test for evidence of cooperative or competitive interaction of bacteria, twelve culture plates were used, of which six were smeared uniformly with 0.5 McFarland suspensions of each of the six test strains to obtain confluent growth (inoculated plate) and the other six were not inoculated (control plates). After left to dry for 5 minutes, 10 µl of 0.5 McFarland standard suspensions of all six test strains were spotted on all culture plates. Agar plates were inspected at day 1, 2, 3, 5 and 7 for bacterial growth on the spots and also around the spots for the inoculated plates. The amount of growth of the spotted strains on the inoculated plates was compared to that on the control plates. Criteria for interpretation were established based on ecological interactions27,28. The nine possible interpretations of the interactions, ranging from mutually beneficial through neutral to spiteful interactions, are outlined in Table S1.
Results
Characteristics of The Patients
Fifty-eight adults [17 control subjects, 21 CRSwNP patients without asthma (CRSwNP−A), and 20 CRSwNP patients with co-morbid asthma (CRSwNP+A)] were enrolled in the study. Table 1 illustrates clinical characteristics of the participants. No significant differences with regard to age or gender were found among the three groups. Atopic status and aspirin intolerance were significantly more prevalent in the CRSwNP+A group, as expected. According to severity of disease based on visual analogue scales for symptoms (VAS), CRSwNP+A patients had remarkably more trouble with nasal congestion and mucus than CRSwNP−A patients (Fig. 1).

Visual analogue scale. The figure shows the symptom profile and symptom severity of the two patient groups. Nasal congestion and mucus scales were significantly higher in asthmatic patients compared to non-asthmatic patients. Statistical analysis was performed using Mann-Whitney test. *p < 0.05; **0.001 < p < 0.01.
Cytokine Patterns in The Nasal Tissue Samples
Nasal polyp (NP) disease in European patients is mostly characterized by an infiltration of eosinophils and expression of high concentrations of ECP. In this study, Th2 related mediators and cytokines (IgE, SE-IgE and IL-5) were significantly increased in NP from asthmatic patients compared with non-asthmatic subjects, as shown in Fig. 2. For example, median total IgE in the CRSwNP−A group was 440.4 kU/L (interquartile ranges IQRs: 101–742.5) compared to 1543 kU/L (IQRs: 640–2321) in the CRSwNP+A group (Table S2). The level of ECP, TNFα, and IL-17 showed no significant differences between groups. Furthermore, the SE-IgE positive group showed notable higher concentrations of IgE (median in the SE-Ig E positive subjects 1307 kU/L vs. 252 kU/L in SE-Ig E negative subjects, p < 0.0001), IL-5 (701 pg/mL vs. 77 pg/mL, p < 0.01), and ECP (29920 μg/L vs. 14045 μg/L, p < 0.01) when compared to the SE-IgE negative group.

Cytokine expression in nasal tissues. The graphs show median, upper and lower quartiles. High IgE, SE-IgE and IL-5 concentrations characterize the inflammation in NP from asthmatic patients. Statistic results were calculated using the t-test for Gaussian-distributed data and the Mann-Whitney test for non-normal distribution. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Microbial Diversity and Relative Abundances of Bacterial Species
Bacterial DNA was detected in all samples. The number of phylotypes varied from 26 to 207 per sample with an average of 113 phylotypes in the control group, 116 phylotypes in the CRSwNP−A group, and 109 phylotypes in the CRSwNP+A group. Regarding the total number of reads of 16S rRNA gene, there was no statistically significant difference among the three groups, indicating that control subjects and CRSwNP patients had about the same total bacterial load. However, the sinonasal microbiota of CRSwNP as a group, regardless of co-morbid asthma, showed significantly decreased bacterial diversity (Shannon H index) and evenness (Pielou’s evenness index), when compared with the control group. Especially, the CRSwNP−A group exhibited significantly reduced bacterial evenness and Shannon’s diversity when compared with the control group. No significant differences were found with regard to Chao-1 species richness indexes in intergroup comparisons (Fig. 3).

Biodiversity indices. The graphs compare diversity indices (Shannon diversity, richness, and evenness) between control subjects and patients with CRSwNP−A and CRSwNP+A. Statistical analysis was performed using Kruskal-Wallis and Dunn’s multiple comparisons test. **0.001 < p < 0.01; ***0.0001 < p < 0.001.
The phylum-level structure of our samples is depicted on Fig. 4. With respect to health and disease status, Phylum Proteobacteria and genus Haemophilus were more abundance in CRSwNP disease than healthy control, while the average abundances of phylum Actinobacteria, Bacteroidetes, and genus Bacteroides were dominant in healthy control subjects when compared with CRSwNP cases. At the species level, Haemophilus influenzae was significantly more abundant in CRSwNP patients than in control subjects. Corynebacterium pseudodiphtheriticum was more prevalent in diseases when compared with controls. Staphylococcus xylosus and Bifidobacterium longum were more prevalent and abundant in the control group than in the CRSwNP group (Fig. 5a).

Bacterial community composition in each subject at the phylum level. C: control subject, P: CRSwNP−A subject, A: CRSwNP+A subject.

Bacterial species that discriminate between groups (a) and among groups (b). The graphs show the relative abundance of discriminative species different between groups. Dots represent relative abundance in each sample. *p < 0.05, **p < 0.01, and ***p < 0.001.
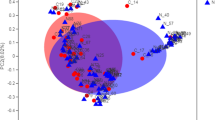
A principal coordinate analysis (PCoA) ordination depicted sinus bacterial beta diversity between samples (Fig. S1). One-way PERMANOVA with Bray-Curtis index detected statistically significant differences of bacterial communities in intergroup comparisons (Holm-Bonferroni sequential corrected p-values < 0.001). A post-hoc pairwise comparison revealed differences in microbial composition between control and CRSwNP+A groups (p < 0.01), control and CRSwNP−A groups (p < 0.01), and between CRSwNP patients (p < 0.03).
Considering the bacterial composition at the phylum level (Fig. 6), Proteobacteria predominated in CRSwNP+A group compared with CRSwNP−A and control group, whereas Actinobacteria were more abundant in the control subjects than in patients with CRSwNP+A. The average abundances of Bacteroidetes were notably higher in control subjects when compared with CRSwNP−A patients. At the genus levels, CRSwNP−A patients carried significantly higher abundance of Corynebacterium (p = 0.04) genera (phylum Actinobacteria) and Geobacter (p < 0.01) genera (phylum Proteobacteria) when compared with CRSwNP+A patients (Fig. S2).

Phylum-level differences between groups. Dot graphs show relative abundance of bacterial phylafrom each sample. Kruskal-Wallis and Dunn’s multiple comparisons were employed for statistical analysis. *p < 0.05; **0.001 < p < 0.01.
At the species level (Fig. 7), Moraxella catarrhalis was more prevalent and abundant in CRSwNP+A subjects, compared to CRSwNP−A subjects, whereas a relative abundance of Geobacter anodireducens/sulfurrreducens and Pelomonas puraquae was noted in CRSwNP−A subjects. All aforementioned species are member of the phylum Proteobacteria. The prevalence of Moraxella catarrhalis, Staphylococcus aureus and Staphylococcus xylosus was significantly lower in CRSwNP−A group when compared with the other groups (p < 0.05). The mean relative abundance of Staphylococcus xylosus was also notably lower in CRSwNP−A group when compared with the other groups. The prevalence and relative abundance of Bifidobacterium longum was higher in healthy group than CRSwNP−A group. For example, Staphylococcus aureus was prevalent in 94% of healthy control subjects, 57% of CRSwNP−A patients, and 90% of CRSwNP+A patients. The average relative abundance of Staphylococcus aureus was 185 in the healthy group, 654 in the CRSwNP−A group, and 145 in the CRSwNP+A group (Fig. S3). Propionibacterium acnes (phylum Actinobacteria) was the most abundant species in the control group, Staphylococcus aureus (phylum Firmicutes) in the CRSwNP−A group and Escherichia coli (phylum Proteobacteria) in the CRSwNP+A group.

Prevalence and mean relative abundance of the most abundant bacterial species in our study. The heat maps in white-to-black and white-to-red depict the prevalence and relative abundance, respectively. The order was arranged based on the contribution to the observed dissimilarity. ns: no statistical significance (p ≥ 0.05); *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Correlations between Cytokine Levels and Relative Abundance of Bacteria
Comparison of cytokine levels with relative abundance of bacteria in the nasal polyps tissues revealed that ECP and IL-5 were negatively correlated with the phylum Actinobacteria (p < 0.05, r = −0.3652, and −0.3760 respectively). The phylum Bacteroidetes correlated positively with IL-5 (p < 0.02, r = 0.3969), but negatively with IL-17 (p < 0.01, r = −0.4632).
At the species level, total IgE and IL-5 were negatively correlated with Geobacter anodireducens/sulfurreducens and Pelomonas puraquae, IL-5 was negatively correlated with Corynebacterium macginleyi, and ECP was negatively correlated with Corynebacterium accolens, C. macginleyi and Streptococcus pneumoniae. In contrast, ECP and IL-5 were positively correlated with E. coli. Staphylococcus aureus and Propionibacterium acnes were not correlated with any cytokine. IL-17 and TNF-α were not correlated with any of the top 20 bacterial species (Table S3). However, no single correlation reached a correlation coefficient of higher than 0.6 (moderate correlation).
Interspecies Interactions of Cultured Bacterial Species
When testing the possible interactions between six strains of five of the most frequently present species, we observed that growth of C. pseudodiptheriticum on the inoculated plate was enhanced by the presence of S. aureus RN6390 and H. influenzae, but retarded by E. coli and inhibited by S. aureus ATCC 29213. P. acnes appeared to be inhibitied by S. aureus (both strains), E. coli and H. influenzae. Furthermore, H. influenzae was inhibited by S. aureus ATCC 29213 and E. coli. Growth of S. aureus RN6390 was inhibited by E. coli.
With respect to growth pattern of the strains used to initially inoculate the plates, C. pseudodiptheriticum swiftly flourished in the proximity of the E. coli inoculation point, whereas growth of P. acnes was inhibited around the inoculation point of S. aureus ATCC 29213. Neither growth inhibition nor growth promotion was noticed between C. pseudodiptheriticum and P. acnes, and between S. aureus ATCC 29213 and E. coli. Figure 8 outlines the observed bacterial growth patterns. Interspecies interplay on the basis of the available information from our in-vitro experiments is depicted in Fig. 9.

Mutual inhibition and growth promotion between different bacterial species that are most abundant in controls and patient groups.

Bacterial in vitro interactions. Red lines represent a negative relationship, green lines represent a positive relationship, and black lines represent a neutral relationship. All bacterial pictures are credited to Centers for Disease Control and Prevention (CDC). The bacterial pictures were taken from Public Health Image Library (https://phil.cdc.gov/phil/home.asp). C. pseudodiptheriticum image ID#: 2126. E. coli image ID#: 10068 photo credit: Janice Harney Carr H. influenzae image ID#: 1947 P. acnes image ID#: 2122 S. aureus image ID#: 11157 photo credit: Janice Harney Carr.
Discussion
The nasal bacterial community has been suggested to play a role in the development and severity of CRSwNP disease29,30. The aim of this study was to establish whether and to what extent there might be differences in the nasal microbiota of healthy control subjects, compared to those of non-asthmatic and asthmatic CRSwNP patients. Subgrouping of the patients into endotypes was based on our previous studies3,6. Indeed, CRSwNP disease can be further classified on the basis of distinct biomarkers (IgE, SE-IgE, and IL-5) that are increased in nasal polyps tissue when there is comorbidity (asthma). Different microbiota might represent different functional communities and invoke an immune response ranging from homeostatic to detrimental inflammatory effects, as such explaining the differences between the different endotypes.
Aberrant bacterial community in CRSwNP disease
This study identifies that the relative abundance of H. influenzae is remarkably higher in CRSwNP cases compared to normal subjects. Combining the experimental data from mice31, the epidemiological data from humans32, and our observation of an association of H. influenzae with CRSwNP, we hypothesize that H. influenzae can initiate and drive inflammatory responses to develop nasal polyposis and therefore it may be hypothesized that elimination of H. influenzae in an acute phase might reduce incidence of NP cases. It might further be assumed that success or failure of the antibiotic treatment of the acute phase infection might drive an ongoing inflammation into resolution or NP formation, respectively.
In addition, the bacterial community in CRSwNP+A patients showed dominance of the phylum Proteobacteria, caused by unexpectedly high numbers of E. coli (Enterobacteriaceae), although not significantly different from the other two groups, whereas S. aureus was predominant in CRSwNP−A patients. Our findings concurred with a previous study that reported strong increase of Enterobacteriaceae in bronchial lavage of patients with asthma compared to control subjects33. When we combined the data of members of the Proteobacteria (i.e. E. coli, H. influenzae, and M. catarrhalis), we could establish a significant increase in relative abundance of these bacteria for patients with CRSwNP+A compared to the control group (Fig. 5b).
Proteobacteria might contribute to the pathogenesis of CRSwNP disease via eicosanoids and related mediators34. The abundance of E. coli in several CRSwNP+A patients and the positive correlation of E. coli with ECP and IL-5 suggest a role of E. coli in severity of type 2 inflammation in CRSwNP patients.
It is possible that E. coli impairs the epithelial barrier and cooperates with putative pathogens for initiation and amplification of type 2 inflammation in CRSwNP disease.
Effect of microbial components on putative pathogens
In the current study, the average relative abundance of S. aureus in CRSwNP−A patients is higher than that in healthy control subjects, but this is not the case for CRSwNP+A patients. The presence of specific IgE to staphylococcal enterotoxins (SE-IgE) is significantly more frequent in the CRSwNP+A compared to the CRSwNP−A and control groups and correlates with disease severity. This observation supports the role of S. aureus and its immune proteome in the persistence of airway disease35,36. Apparently, our findings seem to indicate that the composition of the bacterial community may influence the S. aureus virulence. The presence of large numbers of Actinobacteria (e.g. Corynebacterium spp.) in the environment might shift S. aureus to benign colonization, whereas increased numbers of Proteobacteria (predominantly E. coli) in the milieu might drive S. aureus to exhibit pathogenic behavior. It may be conceivable that bacterial composition and interactions in the community culminate in a functional community that organizes commensal-pathogen interchange. Intraspecific variability cannot be ruled, whereby S. aureus strains present in healthy samples could differ from strains from samples from diseases patients. In one study, a strain of S. aureus inhibited other strains of the same species co-colonizing in nasal cavity37. Various hypotheses were proposed to determine the regulation of S. aureus virulence, of which the global control of accessory gene regulator (agr) is the most promising one38. We also identified strain-level variations of S. aureus, classified by agr, in an independent survey and in-vitro bacterial interaction experiments. S. aureus RN 6390 belonging to agr group I and S. aureus ATCC 29213 agr group II. S. aureus agr group I had a positive impact on C. pseudodiptheriticum, whereas S. aureus agr group II had a negative impact on C. pseudodiptheriticum. Moreover, S. aureus agr group II killed P. acnes in our experiments. A survey of S. aureus agr in higher numbers of patients should be conducted to confirm the possible importance of agr diversity of S. aureus in pathogenesis of CRS diseases.
Corynebacterium species have been implicated as competitors with S. aureus in the nasal niche39,40 or as tempering S. aureus virulence41. One research group observed that C. pseudodiphtheriticum was present more often in non-S. aureus carriers than that in S. aureus carriers42. This group confirmed a competitive interaction between S. aureus and C. pseudodiphtheriticum42, which is not supported by our data. We detected C. pseudodiptheriticum more frequently in the CRSwNP status. In addition, our in-vitro interactions rather indicated a positive cooperation between C. pseudodiptheriticum and putative pathogens, such as E. coli, H. influenzae and S. aureus (agr group I).
Although several studies have described a negative association between S. aureus and S. epidermidis in the nasal cavity40,43, our data did not show competition between S. aureus and S. epidermidis.
Bacterial Associations with Inflammation in CRS
A study using only 7 samples from CRS patients and a not-validated mice model concluded that Corynebacterium tuberculostearicum was a pathogen of CRS44, while another study reported that CRS patients with enriched C. tuberculostearicum colonization in their nose, at the time of endoscopic sinus surgery, showed better surgical outcomes10. However, in our study neither a beneficial nor pathogenic role of C. tuberculostearicum can be concluded. We found that the distribution and relative abundance of C. tuberculostearicum was equivalent among groups. We also observed a negative correlation between eosinophilic activity in the tissue (i.e., ECP and IL-5) and other members of genus Corynebacterium (i.e., C. macginleyi, and C. accolens) that belong to phylum Actinobacteria. According to our previous study, the level of IL-5 is one of the prognostic factors in CRSwNP disease14. These results suggest that members of phylum Actinobacteria, i.e.,Corynebacterium spp. (e.g. C. macginleyi), are associated with a lower degree of eosinophilic inflammation in CRSwNP patients.
Health-associated Microbes
Several bacterial phyla were dominant in healthy individuals. The potentially beneficial microbe in the current study was a member of the phylum Actinobacteria; we found that Propionibacterium acnes was the most abundant microorganism in the healthy condition. Normally, P. acnes is a commensal microbe of adult human skin45. P. acnes, which prefers a lipid-rich anaerobic environment, has been detected in sinonasal cavities of healthy adults9,42,43,46. P. acnes has been described to promote Th1 and Treg cells and relieves atopic dermatitis symptoms47, possibly by the production of short-chain fatty acids that are immunomodulating. Moreover, the principal metabolite of P. acnes, propionic acid, not only reduces inflammation by effects on macrophages48, but also inhibits S. aureus growth49. This argument nevertheless remained unsupported by our pilot study. The levels of propionic acid measured in the sinonasal environment did not reach the minimum inhibitory concentration of S. aureus. Furthermore, P. acnes did not prevent the growth of S. aureus, which in turn killed P. acnes to establish its own growth. These data do indicate that P. acnes is an commensal rather than a defender, although in-vitro data regarding to bacterial interactions cannot be straightforwardly transposed to the in-vivo situation. P. acnes probably possess other factors for its fitness in healthy subjects. Healthy hosts may provide nutrients for P. acnes existence. Health-associated microbes should be further investigated to identify a genuine probiotic for the treatment of CRS disease.
This study provides a blueprint for identifying important microbial influencers of disease that provide information of structure and member of nasal microbiota linked to clinical phenotypes and endotypes such as subgroups of CRSwNP disease (Fig. 10). Our findings highlight the need for more functional studies of bacterial community to determine the role of microbiota in CRS pathogenesis.

Characteristics of bacterial communities for the different groups.
Limitations
Several limitations should be addressed. Although we excluded patients that used steroids systemically, all CRSwNP patients may have taken intranasal steroid until shortly before the biopsy. The effect of steroids on bacterial communities remains unclear, although nasal irrigation with budesonide did not change the proportional abundance of bacteria in CRSwNP patients50. We were concerned about the specificity of molecular techniques, even though it has been shown that the relative abundance of bacterial DNA had a positive correlation with colony-forming unit counts (CFUs)30. Comparing recovered DNA to live colony counting, it appeared that in diseased subjects, Staphylococcus DNA mostly came from living bacteria, whereas in healthy subjects, Staphylococcus DNA was mostly recovered from dead bacteria51. The method used here for microbiota may detect genetic material of non-viable microorganisms that distort the genuine microbial community composition. Moreover, one bacterial cell carries from 1 up to 15 copies 16S rRNA gene copies per genome52. The analysis of 16S rRNA can provide only the information of bacterial presence, but not metabolically active microorganism. The extraction and analysis of metagenomic mRNA (the metatranscriptome) or proteomic or metabolomic research should be further performed to assess the functional and metabolic diversity of microbial communities. Interplays between bacteria in our experiments are performed under in-vitro conditions. Although the tested strains were not directly isolated from the subjects studied, and relevant variations in the metabolites of the microbes may occur, the selected strains were chosen based on investigations in the same patient groups to best represent the situation in the human nose. We tested the interaction of only two species of bacteria. Interactions among the full spectrum of bacteria in-situ conditions may be different. The participation of the third, forth, and more species probably affects those bacterial behavior differently. A complementarily ecological theory would need to explain overarching interactions and patterns for the microbiota. Future research should explore functional parts of bacterial community and their impacts on the nasal mucosa. Investigations of bacterial communities and their impacts on sinonasal immune responses may pave the way for a better understanding of how the microbiota contribute to the pathogenesis of CRS.
Conclusions
This study identifies a difference in nasal microbiota between healthy subjects and phenotypes of CRSwNP patients. Proteobacteria (such as H. influenzae, E.coli, M. catarrhalis) were associated with CRSwNP disease, especially with CRSwNP+A cases, whereas Actinobacteria (such as P. acnes, Corynebacterium spp.) were prevalent in the healthy status. However, P. acnes, a resident commensal bacterium in healthy subjects, does not play the protective role against S. aureus one could expect from its abundance. Furthermore, although S. aureus was found abundant in CRSwNP−A, but not in CRSwNP+A, the presence of SE-IgE in the latter group representing its immunologic fingerprint rather than S. aureus itself is associated with disease severity. The functions of microbiota and their products in health and airway disease should be further elucidated to determine the effective role of microbiota in the pathomechanism of CRS disease.
References
Fokkens, W., Lund, V. & Mullol, J. European Position Paper on, R. & Nasal Polyps, g. European position paper on rhinosinusitis and nasal polyps 2007. Rhinology. Supplement 20, 1–136 (2007).
Batra, P. S., Tong, L. & Citardi, M. J. Analysis of comorbidities and objective parameters in refractory chronic rhinosinusitis. The Laryngoscope 123(Suppl 7), S1–11, https://doi.org/10.1002/lary.24418 (2013).
Tomassen, P. et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. The Journal of allergy and clinical immunology 137, 1449–1456 e1444, https://doi.org/10.1016/j.jaci.2015.12.1324 (2016).
Bachert, C. et al. ICON: chronic rhinosinusitis. The World Allergy Organization journal 7, 25, https://doi.org/10.1186/1939-4551-7-25 (2014).
Ba, L. et al. The association between bacterial colonization and inflammatory pattern in Chinese chronic rhinosinusitis patients with nasal polyps. Allergy 66, 1296–1303, https://doi.org/10.1111/j.1398-9995.2011.02637.x (2011).
Chalermwatanachai, T., Zhang, N., Holtappels, G. & Bachert, C. Association of Mucosal Organisms with Patterns of Inflammation in Chronic Rhinosinusitis. PloS one 10, e0136068, https://doi.org/10.1371/journal.pone.0136068 (2015).
Feazel, L. M., Robertson, C. E., Ramakrishnan, V. R. & Frank, D. N. Microbiome complexity and Staphylococcus aureus in chronic rhinosinusitis. The Laryngoscope 122, 467–472, https://doi.org/10.1002/lary.22398 (2012).
Aurora, R. et al. Contrasting the microbiomes from healthy volunteers and patients with chronic rhinosinusitis. JAMA otolaryngology–head & neck surgery 139, 1328–1338, https://doi.org/10.1001/jamaoto.2013.5465 (2013).
Boase, S. et al. The microbiome of chronic rhinosinusitis: culture, molecular diagnostics and biofilm detection. BMC infectious diseases 13, 210, https://doi.org/10.1186/1471-2334-13-210 (2013).
Ramakrishnan, V. R. et al. Sinus microbiota varies among chronic rhinosinusitis phenotypes and predicts surgical outcome. The Journal of allergy and clinical immunology 136, 334–342 e331, https://doi.org/10.1016/j.jaci.2015.02.008 (2015).
Patou, J. et al. Staphylococcus aureus enterotoxin B, protein A, and lipoteichoic acid stimulations in nasal polyps. The Journal of allergy and clinical immunology 121, 110–115, https://doi.org/10.1016/j.jaci.2007.08.059 (2008).
Corriveau, M. N., Zhang, N., Holtappels, G., Van Roy, N. & Bachert, C. Detection of Staphylococcus aureus in nasal tissue with peptide nucleic acid-fluorescence in situ hybridization. American journal of rhinology & allergy 23, 461–465, https://doi.org/10.2500/ajra.2009.23.3367 (2009).
Bachert, C. et al. Presence of IL-5 protein and IgE antibodies to staphylococcal enterotoxins in nasal polyps is associated with comorbid asthma. The Journal of allergy and clinical immunology 126, 962–968, 968 e961–966, https://doi.org/10.1016/j.jaci.2010.07.007 (2010).
Van Zele, T., Holtappels, G., Gevaert, P. & Bachert, C. Differences in initial immunoprofiles between recurrent and nonrecurrent chronic rhinosinusitis with nasal polyps. American journal of rhinology & allergy 28, 192–198, https://doi.org/10.2500/ajra.2014.28.4033 (2014).
Lee, C. W., Lee, B. J., Yoo, S. H. & Yi, J. S. Relationship between positive bacterial culture in maxillary sinus and surgical outcomes in chronic rhinosinusitis with nasal polyps. Auris, nasus, larynx 41, 446–449, https://doi.org/10.1016/j.anl.2014.05.010 (2014).
Biswas, K., Hoggard, M., Jain, R., Taylor, M. W. & Douglas, R. G. The nasal microbiota in health and disease: variation within and between subjects. Frontiers in microbiology 9, 134, https://doi.org/10.3389/fmicb.2015.00134 (2015).
Cope, E. K., Goldberg, A. N., Pletcher, S. D. & Lynch, S. V. Compositionally and functionally distinct sinus microbiota in chronic rhinosinusitis patients have immunological and clinically divergent consequences. Microbiome 5, 53, https://doi.org/10.1186/s40168-017-0266-6 (2017).
Newton, J. R. & Ah-See, K. W. A review of nasal polyposis. Therapeutics and Clinical Risk Management 4, 507–512 (2008).
Fokkens, W. J. et al. EPOS 2012: European position paper on rhinosinusitis and nasal polyps 2012. A summary for otorhinolaryngologists. Rhinology 50, 1–12, https://doi.org/10.4193/Rhino50E2 (2012).
Camarinha-Silva, A. et al. Comparing the anterior nare bacterial community of two discrete human populations using Illumina amplicon sequencing. Environmental microbiology 16, 2939–2952, https://doi.org/10.1111/1462-2920.12362 (2014).
Pruesse, E. et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35, 7188–7196, https://doi.org/10.1093/nar/gkm864 (2007).
McMurdie, P. J. & Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PloS one 8, e61217, https://doi.org/10.1371/journal.pone.0061217 (2013).
R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2014).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73, 5261–5267, https://doi.org/10.1128/aem.00062-07 (2007).
Verstraelen, H. et al. Characterisation of the human uterine microbiome in non-pregnant women through deep sequencing of the V1-2 region of the 16S rRNA gene. PeerJ 4, e1602, https://doi.org/10.7717/peerj.1602 (2016).
Øyvind Hammer, D. A. T. Ha. P. D. R. PAST: Paleontological statistics software package for education and data analysis. Palaeontologia Electronica 4, 9 (2001).
Hamilton, W. D. The genetical evolution of social behaviour. I. Journal of theoretical biology 7, 1–16 (1964).
Faust, K. & Raes, J. Microbial interactions: from networks to models. Nature reviews. Microbiology 10, 538–550, https://doi.org/10.1038/nrmicro2832 (2012).
Chalermwatanachai, T., Velasquez, L. C. & Bachert, C. The microbiome of the upper airways: focus on chronic rhinosinusitis. The World Allergy Organization journal 8, 3, https://doi.org/10.1186/s40413-014-0048-6 (2015).
Ramakrishnan, V. R., Hauser, L. J. & Frank, D. N. The sinonasal bacterial microbiome in health and disease. Current opinion in otolaryngology & head and neck surgery 24, 20–25, https://doi.org/10.1097/moo.0000000000000221 (2016).
McCann, J. R., Mason, S. N., St Auten, R. L., Geme, J. W. 3rd & Seed, P. C. Early-Life Intranasal Colonization with Nontypeable Haemophilus influenzae Exacerbates Juvenile Airway Disease in Mice. Infect Immun 84, 2022–2030, https://doi.org/10.1128/iai.01539-15 (2016).
Van Bruaene, N. et al. T-cell regulation in chronic paranasal sinus disease. The Journal of allergy and clinical immunology 121, 1435–1441, 1441 e1431-1433, https://doi.org/10.1016/j.jaci.2008.02.018 (2008).
Marri, P. R., Stern, D. A., Wright, A. L., Billheimer, D. & Martinez, F. D. Asthma-associated differences in microbial composition of induced sputum. The Journal of allergy and clinical immunology 131, 346–352 e341-343, https://doi.org/10.1016/j.jaci.2012.11.013 (2013).
Van Crombruggen, K., Zhang, N., Gevaert, P., Tomassen, P. & Bachert, C. Pathogenesis of chronic rhinosinusitis: inflammation. The Journal of allergy and clinical immunology 128, 728–732, https://doi.org/10.1016/j.jaci.2011.07.049 (2011).
Bachert, C. & Zhang, N. Chronic rhinosinusitis and asthma: novel understanding of the role of IgE ‘above atopy’. Journal of internal medicine 272, 133–143, https://doi.org/10.1111/j.1365-2796.2012.02559.x (2012).
Bachert, C. et al. Specific IgE against Staphylococcus aureus enterotoxins: an independent risk factor for asthma. The Journal of allergy and clinical immunology 130, 376–381 e378, https://doi.org/10.1016/j.jaci.2012.05.012 (2012).
Barbagelata, M. S. et al. Auxotrophic mutant of Staphylococcus aureus interferes with nasal colonization by the wild type. Microbes and infection 13, 1081–1090, https://doi.org/10.1016/j.micinf.2011.06.010 (2011).
Bronner, S., Monteil, H. & Prevost, G. Regulation of virulence determinants in Staphylococcus aureus: complexity and applications. FEMS microbiology reviews 28, 183–200, https://doi.org/10.1016/j.femsre.2003.09.003 (2004).
Uehara, Y. et al. Bacterial interference among nasal inhabitants: eradication of Staphylococcus aureus from nasal cavities by artificial implantation of Corynebacterium sp. J Hosp Infect 44, https://doi.org/10.1053/jhin.1999.0680 (2000).
Lina, G. et al. Bacterial Competition for Human Nasal Cavity Colonization: Role of Staphylococcal agr Alleles. Applied and Environmental Microbiology 69, 18–23, https://doi.org/10.1128/aem.69.1.18-23.2003 (2003).
Ramsey, M. M., Freire, M. O., Gabrilska, R. A., Rumbaugh, K. P. & Lemon, K. P. Staphylococcus aureus Shifts toward Commensalism in Response to Corynebacterium Species. Frontiers in microbiology 7, https://doi.org/10.3389/fmicb.2016.01230 (2016).
Yan, M. et al. Nasal microenvironments and interspecific interactions influence nasal microbiota complexity and S. aureus carriage. Cell host & microbe 14, 631–640, https://doi.org/10.1016/j.chom.2013.11.005 (2013).
Frank, D. N. et al. The human nasal microbiota and Staphylococcus aureus carriage. PloS one 5, e10598, https://doi.org/10.1371/journal.pone.0010598 (2010).
Abreu, N. A. et al. Sinus Microbiome Diversity Depletion and Corynebacterium tuberculostearicum Enrichment Mediates Rhinosinusitis. Science translational medicine 4, 151ra124 (2012).
Costello, E. K. et al. Bacterial Community Variation in Human Body Habitats Across Space and Time. Science 326, 1694–1697 (2009).
Ramakrishnan, V. R. et al. The microbiome of the middle meatus in healthy adults. PloS one 8, e85507, https://doi.org/10.1371/journal.pone.0085507 (2013).
Kitagawa, H. et al. Propionibacterium acnes vaccination induces regulatory T cells and Th1 immune responses and improves mouse atopic dermatitis. Experimental dermatology 20, 157–158, https://doi.org/10.1111/j.1600-0625.2010.01180.x (2011).
Al-Lahham, S. et al. Propionic acid affects immune status and metabolism in adipose tissue from overweight subjects. European journal of clinical investigation 42, 357–364, https://doi.org/10.1111/j.1365-2362.2011.02590.x (2012).
Wang, Y. et al. Propionic acid and its esterified derivative suppress the growth of methicillin-resistant Staphylococcus aureus USA300. Beneficial microbes 5, 161–168, https://doi.org/10.3920/bm2013.0031 (2014).
Liu, C. M. et al. Impact of saline irrigation and topical corticosteroids on the postsurgical sinonasal microbiota. International forum of allergy & rhinology 5, 185–190, https://doi.org/10.1002/alr.21467 (2015).
Nakatsuji, T. et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Science translational medicine 9, https://doi.org/10.1126/scitranslmed.aah4680 (2017).
Klappenbach, J. A., Saxman, P. R., Cole, J. R. & Schmidt, T. M. rrndb: the Ribosomal RNA Operon Copy Number Database. Nucleic Acids Research 29, 181–184 (2001).
Acknowledgements
C. Bachert is supported by grants from the European Commission’s Seventh Framework programme under grant agreement no. 260895 (PREDICTA); from the Flemish Scientific Research Board, FWO, No. A12/5-HB- KH3, by the Global Allergy and Asthma European Network (GA2LEN) project G.0436.04, 3G.0489, G.0642.10N; and by the Interuniversity Attraction Poles Program–Belgian State Belgian Science Policy, No. IAP P7/30. C. Thanit is supported by scholarship from the Royal Thai Government. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conception or design of the experiments: T.C., M.V. and C.B. Performed the experiments: T.C., R.V., G.H., T.L., R.J. and D.P. Data analysis and interpretation: T.C., R.V., G.H. and F.M.K. Drafting the article: T.C. Critical revision of the article T.V.W., M.V., T.V.Z. and C.B. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chalermwatanachai, T., Vilchez-Vargas, R., Holtappels, G. et al. Chronic rhinosinusitis with nasal polyps is characterized by dysbacteriosis of the nasal microbiota. Sci Rep 8, 7926 (2018). https://doi.org/10.1038/s41598-018-26327-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-26327-2
This article is cited by
-
The cyclooxygenase-2 upregulation mediates production of PGE2 autacoid to positively regulate interleukin-6 secretion in chronic rhinosinusitis with nasal polyps and polyp-derived fibroblasts
Scientific Reports (2024)
-
Activation of the hedgehog signaling pathway is associated with the promotion of cell proliferation and epithelial–mesenchymal transition in chronic rhinosinusitis with nasal polyps
European Archives of Oto-Rhino-Laryngology (2023)
-
A Review on the Nasal Microbiome and Various Disease Conditions for Newer Approaches to Treatments
Indian Journal of Otolaryngology and Head & Neck Surgery (2023)
-
Microbiota dysbiosis in odontogenic rhinosinusitis and its association with anaerobic bacteria
Scientific Reports (2022)
-
Eosinophils in the Field of Nasal Polyposis: Towards a Better Understanding of Biologic Therapies
Clinical Reviews in Allergy & Immunology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
