
Abstract
Understanding blood group antigen binding preferences for C-type lectin receptors holds promise for modulating immune responses, since several Gram-negative bacteria express blood group antigens as molecular mimicry to evade immune responses. Herein, we report the synthesis of ABO blood group antigen active tri and disaccharides to investigate the binding specificity with various C-type lectin receptors using glycan microarray. The results of binding preferences show that distinct glycosylation on the galactose and fucose motifs are key for C-type lectin receptor binding and that these interactions occur in a Ca2+-dependent fashion.
Similar content being viewed by others

Introduction
Many pathogens display blood group antigens and their molecular mimics on their cell surfaces to escape the immune system of the host1,2,3. For example, Escherichia coli O86:K2:H2 and O86:K61:B7 display B-antigen oligosaccharides core (α-D-Gal-(1-3)-[α-L-Fuc-(1-2)]β-D-Gal-(1-3)-α-D-GalNac-(1-3)-GalNAc) in the lipopolysaccharide part and cause diarrhea in children4. Similarly, E. coli O90, O127 and O128 also express human blood group antigen epitopes and Lewisx on their cell surfaces to evade host immune responses5. In 1960s, Springer and co-workers reported that several gram-negative bacteria display blood group determinants6. Moreover, cancer-associated carbohydrate antigens such as Globo-H carry structural features of O-antigen7,8. ABO(H) antigens are composed of distinct carbohydrate structures that differ only in their terminal monosaccharide structures. Animal lectins such as siglecs, galectins and C-type lectins have been shown to mediate pathogen recognition and subsequent immune responses9,10,11,12,13,14. In addition, galectins binding preference to blood group antigens was described15,16. Given the importance of C-type lectin receptors as microbial and cancer-associated pattern recognition receptors, herein, we report the synthesis of ABO active tri- and disaccharides that were then printed as glycan microarrays to study C-type lectin receptor (CLR) binding specificities. CLR are defined as such by their Ca2+-dependent binding to their antigens of selective carbohydrate-protein interactions17. Mg2+ are also known to have stabilizing effect albeit weaker than the Ca2+ in CLR binding18. Using four CLRs as CLR-Fc fusion proteins, we investigate glycan-protein interactions in a carbohydrate array format. The selected CLR-hFc fusion proteins were human DC-SIGN and three murine CLRs (Mincle-hFc, MGL-1-hFc and SIGNR3-hFc). Based on the binding preferences, we have rationalized the C-type lectin selectivity.
Results and Discussion
Synthesis of ABO active glycans
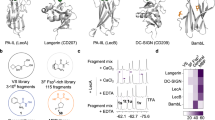
The ABO active glycan and fucosyl derivative (1-4) were synthesized from fully protected tri and disaccharides (5, 6, 11 and 13), respectively (Fig. 1). The assembly of A and B-antigens (GalNAcα(1-3)(Fucα(1-2))GalβPEGNH2 (1) and Galα(1-3)(Fucα(1-2))GalβPEGNH2) (2) involved the thiogalactoside building block 7 and 8, and of reducing end sugar 9a/b. In contrast, the assembly of 3 (O-antigen, Fucα(1-2)GalβPEGNH2) and 4 (Fucα(1-3)GlcNAcβPEGNH2) involved the assembly of thiofucoside donor 10 and reducing end sugar 4,6-benzilidine-galactoside 12a and glucosamine acceptor 14, respectively. The thiogalactoside building block 7 and thiofucoside donor 10 were synthesized as previously described19,20,21,22. The donor 8 was synthesized from a known thiogalactoside donor 15, which was synthesized from D-galactose in 4 steps, as described22. Benzylation of the C-2 and C-3 hydroxyl function of 15 provided the desired thiogalactoside building block 8 (Fig. 2). The synthesis of thiogalactose acceptor 9a/b involved the C-3 PMB protection of 15 by using PMBCl in the presence of stannylene acetate formation, followed by C-2 benzoylation and glycosylation with 2-azidoethoxyethanol linker yielded 12, and the cleavage of C-3 PMB ether protection in the presence of 2,3-dichloro-5,6,-dicyano-1,4-benzoquinone (DDQ) yielded acceptor 9a and in situ protection of Cbz by reduction of linker azide yielded 9b (Fig. 2). The 4,6-benzilidine-NHTroc-glucosamine reducing end sugar was synthesized from 1823 by glycosylation with the linker to yield 14. With all building blocks in hand, we carried out glycosylation using standard NIS/TfOH mediated condition at −40 °C. The thiogalactoside donor 8 and 2-azido-2-deoxy-thiogalacoside donor 7 was glycosylated with 9a/b to obtain disaccharides 19 and 20 (Fig. 3A). To control the stereochemistry of GalN3-α(1-3)-Gal (19) and Gal-α(1-3)-Gal (20), the fucosyl building block 10 was incorporated after the assembly of the disaccharides. Finally, the global deprotection and acetylation of galactosamine resulted in the fully deprotected A- and B-group trisaccharides (1-2) in moderate yields (Fig. 3A). Similarly, glycosylation between thiofucoside donor 10 with acceptor 12a and 14 yielded fully protected disaccharides 11 and 13, respectively. Finally, the global deprotection, followed by acetylation of glucosamine resulted in the disaccharides 3 and 4 in reasonable yields (Fig. 3B).

Retrosynthesis of compounds 1-4.

Synthesis of building blocks: (a) PMBCl, dibutyltinoxide, Bu2SnO, TBAI, toluene, 104 °C; (b) BnBr, NaH, DMF, 0 °C; (c) BzCl/Pyridine, 0 °C; (d) 2-azidoethoxyethanol, NIS/TfOH, DCM, −40 °C, 4 Å molecular sieves (e) DDQ, DCM/Water [18:1 (v/v)], (f) Zn, AcOH, and CbzCl, NaHCO3, THF/H2O, 0 °C.

(A and B). Synthesis of 1-4 glycans: (a) NIS/TfOH, DCM, −40 to −20 °C, 4 Å molecular sieves 7/8 with 9a/b; 12a/14/21/22 with 10; (b) NaOMe, MeOH; (c) HCOOH, Pd(OH)2/H2, MeOH; (d) Zn, THF, AcOH, Ac2O(v/v, 3:2:1); (e) LiOH.H2O, 1,4-dioxane:H2O (v/v, 1:1), 80 °C.
Microarray analysis
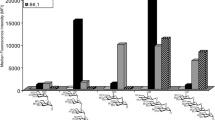
Next, the synthetic 1-4 were printed onto epoxide-functionalized microarray slides at 50 μM in replicates of four, as described in the experimental section24,25. CLR-hFc fusion proteins were incubated on the slide at 20 ng μl−1 (DC-SIGN, SIGNR3, Mincle and MGL-1)26,27 in optimized conditions of 50 mM HEPES buffer with 5 mM CaCl2, 5 mM MgCl2, 0.005% Tween-20 and 1% ovalbumin, followed by the secondary antibody (Cy3-anti-human IgG). During all binding and washing steps divalent cations were maintained. In control blocks, divalent cations were omitted from all binding and washing steps. Slides were scanned and the binding was determined by the fluorescence intensity, as described in the experimental section (Fig. 4). In addition, binding to 1-4 on the microarray was analyzed using IVIG (at 1 µg μl−1; human IgG pool, known to bind many glycans) and the biotinylated plant lectin PNA at 20 ng μl−1. As expected, the PNA lectin, a galactose specific plant lectin, bound to 2-3, and the human IgG pool bound to ABO antigens confirming the conjugation of glycans to the slides (Fig. S6). Furthermore, all four C-type lectins displayed binding preference to ABO active saccharides. However, they exhibited different binding preferences, clearly indicating the differences in structure-binding relationships. Human DC-SIGN, a fucose-specific lectin28,29,30,31,32 bound to all four glycans with statistically significant greater responsiveness towards 1, suggesting that galactose residue may also be important for optimal binding preferences. In contrast, the high-mannose and fucose glycan specific SIGNR3 lectin33,34,35,36 showed weak binding of 4 compared to 1-3, highlighting that the structural-activity relationship of SIGNR3 is different from DC-SIGN. On the other hand, MGL-137 showed preferential binding towards A-antigen active trisaccharide compared to compound 2-4 which were also recognized38,39,40. Finally, Mincle lectin, which is known to bind trehalose derivatives41, surprisingly displayed some binding preference towards 3 compared to the other glycans. The disparity in the binding preference clearly indicates the galactose/galactosamine and fucose residues of 1-3 significantly influence the binding pattern of the CLR-Fc fusion proteins. In order to analyze the Ca2+ dependency of the CLR-hFc interactions with 1-4, we screened the binding preferences in the absence of Ca2+ (and Mg2+) ions. As expected, the binding affinity of specific antigen active molecules to the CLR-hFc fusion proteins significantly decreased. Some of these lectins had been previously examined on glycan microarrays of the Consortium for Functional Glycomics (CFG; http://www.functionalglycomics.org/static/consortium/resources/resourcecoreh.shtml), including against blood group antigens. DC-SIGN and MGL1 exhibited somewhat different binding patterns, while SIGNR3 had not been examined. Binding patterns may be affected by ABO semi-synthetic oligosaccharides vs full length oligosaccharides microarray analysis, differences in the glycan linker, conjugation chemistry to the slides, microarray slide coat, and lectin binding detection method10a. Altogether, the structure-activity data shows that blood group antigens bind to C-type lectins, yet with different preferences. Future studies could address binding affinities and avidity while examining multimeric CLR units to resemble the native receptors function. Furthermore, these results provide the basis for understanding of how pathogens may target CLRs by molecular mimicry to evade immune responses.

Microarray analysis of compounds 1-4 with C-type lectin-Fc fusion proteins (DC-SIGN, SIGNR3, Mincle and MGL1). Mean ± SD of four replicates per measurement.
Conclusions
In conclusion, we have synthesized a series of blood group antigens and immobilized them on glycan microarray slides to determine C-type lectin receptor binding preferences. We have demonstrated that galactose and fucose moieties in 1-4 significantly influence the binding preferences of specific C-type lectins, in a Ca2+-dependent manner. Overall, comparison of the C-type lectin binding patterns allows for further understanding of the basic differences in their preferential recognition of blood group antigens that constitutes a valuable tool for interfering with these interactions.
Methods
General procedure for the production of the CLR-Fc library
The general procedure for the production of human and mouse CLR-Fc fusion protein library has been described previously24,42. The following primers (Table 1) were used for PCR amplification of cDNA fragments encoding the extracellular part of the respective C-type lectin receptor (CLR).
The cDNA fragments were cloned into the pDrive cloning vector (Qiagen) and further ligated into the pFuse-hIgG1-Fc expression vector (InvivoGen). Next, the CLR-hFc encoding vectors were transiently transfected using the FreeStyle Max CHO-S Expression System (Life Technologies). The cell supernatant containing the CLR-hFc fusion proteins was collected and the CLR-hFc fusion proteins were purified using HiTrap Protein G HP columns (GE Healthcare). Identity and purity of the CLR-Fc fusion proteins were confirmed by SDS-PAGE with subsequent Coomassie stain as well as Western Blot. Concentration determination was performed using the Micro BCA Protein Assay Kit (Thermo Scientific).
Microarray fabrication
Arrays were fabricated with NanoPrint LM-60 Microarray Printer (Arrayit) on epoxide-derivatized slides (PolyAn 2D) with 16 sub-array blocks on each slide. Glycoconjugates were distributed into 384-well source plates using 4 replicate wells per sample and 8 µl per well (Version Vr.). Each glycoconjugate was prepared at 50 µM in an optimized print buffer (300 mM phosphate buffer, pH 8.4 supplemented with 0.005% Tween-20). To monitor printing quality AlexaFlour-555-Hydraside (Invitrogen, at 2 ng/µl in 178 mM phosphate buffer, pH 5.5) was used for each printing run. The arrays were printed with four SMP3 pins (5 µm tip, 0.25 µl sample channel, ~100 µm spot diameter; Arrayit), with spot to spot spacing of 275 µm. The humidity level in the arraying chamber was maintained at about 70% during printing. Printed slides were left on arrayer deck over-night, allowing humidity to drop to ambient levels (40–45%). Next, slides were packed, vacuum-sealed and stored at room temperature (RT) until used.
Microarray binding assay
Slides were developed and analyzed as previously described24,25 with some modifications. Slides were rehydrated with dH2O and incubated for 30 min in a staining dish with 50 °C pre-warmed ethanolamine (0.05 M) in Tris-HCl (0.1 M, pH 9.0) to block the remaining reactive epoxy groups on the slide surface, then washed with 50 °C pre-warmed dH2O. Slides were centrifuged at 200 × g for three min then fitted with ProPlate™ Multi-Array 16-well slide module (Grace Bio-lab P37001) to divide into the sub-arrays (blocks). Slides were washed with washing buffer (50 mM HEPES pH 7, 5 mM CaCl2, 5 mM MgCl2, 0.005% Tween 20), aspirated and blocked with 200 µl/sub-array of Blocking buffer (50 Mm HEPES pH 7, 5 mM CaCl2, 5 mM MgCl2, 0.005% Tween 20 and 1% w/v Ovalbumin) for 1 hour at RT with gentle shaking. Next, the blocking buffer was aspirated and 100 µl/block of C-type lectins (20 ng/µl), plant lectin Bio-PNA (20 ng/µl) and IVIG GammaGard 1 mg/ml (all diluted blocking buffer, except Bio-PNA that was diluted in blocking buffer C: 50 Mm HEPES pH 7, 20 mM CaCl2, 5 mM MnCl2, 0.005% Tween 20 and 1% w/v Ovalbumin) were incubated with gentle shaking for 2 hours at RT. Slides were washed 4 times with washing buffer, then with washing buffer without Tween-20 for 2 min. Bound antibodies were detected by incubating with secondary detection diluted in washing buffer, 200 µl/block at RT for 1 hour: Cy3-anti-human IgG, H + L (1.5 µg/ml) or Cy3-SA (0.75 µg/ml). Slides were washed 4 times with washing buffer, then with washing buffer for 10 min followed by removal from ProPlate™ Multi-Array slide module and immediately dipping in a staining dish with dH2O supplemented with 5 mM CaCl2, 5 mM MgCl2 for 10 min with shaking. Slides then were centrifuged at 200 × g for 3 min and the dry slides immediately scanned.
Array slide processing
Processed slides were scanned and analyzed described4,5 at 10 μm resolution with a Genepix 4000B microarray scanner (Molecular Devices) using 350 gain. Image analysis was carried out with Genepix Pro 6.0 analysis software (Molecular Devices). Spots were defined as circular features with a variable radius as determined by the Genepix scanning software. Local background subtraction was performed.
References
Stowell, S. R. et al. Innate immune lectins kill bacteria expressing blood group antigen. Nat. Med. 16, 295–301 (2010).
Rowe, J. A. et al. Blood group O protects against severe Plasmodium falciparum malaria through the mechanism of reduced rosetting. Proc. Natl. Acad. Sci. 104, 17471–17476 (2007).
Li, M. et al. Identification of a New R 1, 2-Fucosyltransferase Involved in O-Antigen Biosynthesis of Escherichia coli O86: B7 and Formation of H-Type 3 Blood Group Antigen. 11590–11597 (2008).
Mani, R., Udgaonkar., U. & Pawar, S. Study of enteropathogenic Escherichia coli(EPEC) diarrhoea in children. Indian J Pathol Microbiol. 46, 118–120 (2003).
Stenutz, R., Weintraub, A. & Widmalm, G. The structures of Escherichia coli O-polysaccharide antigens. FEMS Microbiol. Rev. 30, 382–403 (2006).
Springer, G. F. & Horton, R. E. Blood group isoantibody stimulation in man by feeding blood group-active bacteria. J. Clin. Invest. 48, 1280–1291 (1969).
Huang, C. Y. et al. Carbohydrate microarray for profiling the antibodies interacting with Globo H tumor antigen. Proc Natl Acad Sci USA 103, 15–20 (2006).
Jeon, I., Iyer, K. & Danishefsky, S. J. A practical total synthesis of globo-H for use in anticancer vaccines. J. Org. Chem. 74, 8452–8455 (2009).
Cash, H. L., Whitham, C. V., Behrendt, C. L. & Hooper, L. V. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 313, 1126–1130 (2006).
Figdor, C. G., van Kooyk, Y. & Adema, G. J. C-Type Lectin receptors on Dendritic cells and Langerhans Cells. Nat. Rev. Immunol. 2, 77–84 (2002).
van Kooyk, Y. & Rabinovich, G. A. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 9, 593–601 (2008).
Geijtenbeek, T. B. H. & Gringhuis, S. I. Signalling through C-type lectin receptors: Shaping immune responses. Nat. Rev. Immunol. 9, 465–479 (2009).
Mayer, S., Raulf, M. K. & Lepenies, B. C-type lectins: their network and roles in pathogen recognition and immunity. Histochem. Cell Biol. 147, 223–237 (2017).
Crocker, P. R., Paulson, J. C. & Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 7, 255–266 (2007).
Horlacher, T. et al. Determination of carbohydrate-binding preferences of human galectins with carbohydrate microarrays. ChemBioChem 11, 1563–1573 (2010).
Stowell, S. R. et al. Galectin-1, -2, and -3 exhibit differential recognition of sialylated glycans and blood group antigens. J. Biol. Chem. 283, 10109–10123 (2008).
Cummings R. D., McEver, R. P. C-type lectins. Essentials of Glycobiology. 3rd edition, Cold Spring Harbor Laboratory Press; 2015-2017, Chapter 24 (2017).
Hag, S., Kubo, T., Kurata, S., Kobayashi, A. & Natori, S. Purification, characterization, and cDNA cloning of a galactose-specific C-type lectins from Drosophila melanogaster. J. Biol. Chem. 271(33), 20213–20218 (1996).
Ingle, A. B., Chao, C. S., Hung, W. C. & Mong, K. K. T. Chemical Synthesis of the O-Antigen Repeating Unit of Escherichia coli O86 by an N-Formylmorpholine-Modulated One-Pot Glycosylation Strategy. Asian J. Org. Chem. 3, 870–876 (2014).
Kalikanda, J. & Li, Z. Study of the stereoselectivity of 2-azido-2-deoxygalactosyl donors: Remote protecting group effects and temperature dependency. J. Org. Chem. 76, 5207–5218 (2011).
Hsu, C. H. et al. Highly alpha-selective sialyl phosphate donors for efficient preparation of natural sialosides. Chem. - A Eur. J. 16, 1754–1760 (2010).
Yadav, R., Leviatan Ben-Arye, S., Subramani, B., Padler-Karavani, V. & Kikkeri, R. Screening of Neu5Acα(2–6)gal isomer preferences of siglecs with a sialic acid microarray. Org. Biomol. Chem. 14, 10812–10815 (2016).
Wang, Z., Zhou, L., El-Boubbou, K., Ye, X. S. & Huang, X. Multi-component one-pot synthesis of the tumor-associated carbohydrate antigen globo-H based on preactivation of thioglycosyl donors. J. Org. Chem. 72, 6409–6420 (2007).
Padler-Karavani, V. et al. Cross-comparison of protein recognition of sialic acid diversity on two novel sialoglycan microarrays. J. Biol. Chem. 287, 22593–22608 (2012).
Leviatan Ben-Arye, S., Yu, H., Chen, X. & Padler-Karavani, V. Profiling Anti-Neu5Gc IgG in Human Sera with a Sialoglycan Microarray Assay. J. Vis. Exp. 1–10, https://doi.org/10.3791/56094 (2017).
Maglinao, M. et al. A platform to screen for C-type lectin receptor-binding carbohydrates and their potential for cell-specific targeting and immune modulation. J. Control. Release 175, 36–42 (2014).
Lepenies, B., Lee, J. & Sonkaria, S. Targeting C-type lectin receptors with multivalent carbohydrate ligands. Adv. Drug Deliv. Rev. 65, 1271–1281 (2013).
van Die, I. et al. The dendritic cell-specific C-type lectin DC-SIGN is a receptor for Schistosoma mansoni egg antigens and recognizes the glycan antigen Lewis x. Glycobiology 13, 471–478 (2003).
Guo, Y. et al. Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nat. Struct. Mol. Biol. 11, 591–598 (2004).
Singh, S. K. et al. Targeting glycan modified OVA to murine DC-SIGN transgenic dendritic cells enhances MHC class I and II presentation. Mol. Immunol. 47, 164–174 (2009).
Guzzi, C. et al. Insights into molecular recognition of Lewis(X) mimics by DC-SIGN using NMR and molecular modelling. Org. Biomol. Chem. 9, 7705–7712 (2011).
Adachi, Y. et al. Characterization of β -Glucan Recognition Site on C-Type Lectin, Dectin 1 Characterization of β-Glucan Recognition Site on C-Type Lectin, Dectin 1. Infect. Immun. 72, 4159–4171 (2004).
Galustian, C. et al. High and low affinity carbohydrate ligands revealed for murine SIGN-R1 by carbohydrate array and cell binding approaches, and differing specificities for SIGN-R3 and langerin. Int. Immunol. 16, 853–866 (2004).
Powlesland, A. S. et al. Widely divergent biochemical properties of the complete set of mouse DC-SIGN-related proteins. J. Biol. Chem. 281, 20440–20449 (2006).
Bavireddi, H. et al. Understanding carbohydrate–protein interactions using homologous supramolecular chiral Ru(II)-glyconanoclusters. Nanoscale 8, 19696–19702 (2016).
Brzezicka, K. et al. Influence of Core β-1,2-Xylosylation on Glycoprotein Recognition by Murine C-type Lectin Receptors and Its Impact on Dendritic Cell Targeting. ACS Chem. Biol. 11, 2347–2356 (2016).
Artigas, G. et al. Glycopeptides as Targets for Dendritic Cells: Exploring MUC1 Glycopeptides Binding Profile toward Macrophage Galactose-Type Lectin (MGL) Orthologs. J. Med. Chem. 60, 9012–9021 (2017).
Singh, S. K. et al. Characterization of murine MGL1 and MGL2 C-type lectins: Distinct glycan specificities and tumor binding properties. Mol. Immunol. 46, 1240–1249 (2009).
Eriksson, M. et al. Biological evaluation of multivalent Lewis X-MGL-1 interactions. ChemBioChem 15, 844–851 (2014).
Streng-Ouwehand, I. et al. Glycan modification of antigen alters its intracellular routing in dendritic cells, promoting priming of T cells. Elife 5, 1–21 (2016).
Jegouzo, S. A. F. et al. Defining the conformation of human mincle that interacts with mycobacterial trehalose dimycolate. Glycobiology 24, 1291–1300 (2014).
Mayer, S. et al. C-Type Lectin Receptor (CLR)-Fc Fusion Proteins As Tools to Screen for Novel CLR/Bacteria Interactions: An Exemplary Study on Preselected Campylobacter jejuni Isolates. Front. Immunol. 9, 213 (2018).
Acknowledgements
Financial support from the IISER, Pune, Max-Planck partner group and DST (Grant No. SB/S1/C-46/2014) is gratefully acknowledged (to R.K). This work was also supported by the European Union H2020 Program grants (ERC-2016-STG-716220), and by the Israeli National Nanotechnology Initiative and Helmsley Charitable Trust for a Focal Technology Area on Nanomedicines for Personalized Theranostics (to V.P.-K.). J.T.M. and B.L. acknowledge funding from the European Union’s Horizon 2020 research and innovation program (Marie Sklodowska-Curie grant agreement No. 642870, ETN-Immunoshape). Previously, funding from the Collaborative Research Center (SFB) 765 was crucial for the research program of B.L. We thank Maha Maglinao, Magdalena Eriksson, Stephanie Zimmermann, and Timo Johannssen for help with the generation of the CLR-Fc fusion proteins.
Author information
Authors and Affiliations
Contributions
R.K., V.P.K., and B.L. conceived and directed the project; C.D.S., P.J., and B.S. designed and synthesized the antigens; S.Y., and S.L.B.A. performed microarray studies; J.T.M. produced the C-type lectin-Fc fusion proteins.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shanthamurthy, C.D., Jain, P., Yehuda, S. et al. ABO Antigens Active Tri- and Disaccharides Microarray to Evaluate C-type Lectin Receptor Binding Preferences. Sci Rep 8, 6603 (2018). https://doi.org/10.1038/s41598-018-24333-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-24333-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
