
Abstract
Inflammation is an established contributor to disease and the NLRP3 inflammasome is emerging as a potential therapeutic target. A number of small molecule inhibitors of the NLRP3 pathway have been described. Here we analysed the most promising of these inhibitor classes side by side to assess relative potency and selectivity for their respective putative targets. Assessed using ASC inflammasome-speck formation, and release of IL-1β, in both human monocyte/macrophage THP1 cells and in primary mouse microglia, we compared the relative potency and selectivity of P2X7 inhibitors, inflammasome inhibitors (diarylsulfonylurea vs. the NBC series), and caspase-1 inhibitors. In doing so we are now able to provide a well characterised small molecule tool kit for interrogating and validating inflammasome-dependent responses with a range of nanomolar potency inhibitors against established points in the inflammasome pathway.
Similar content being viewed by others

Introduction
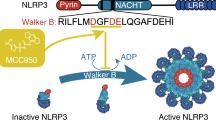
Inflammation is a protective host response to infection, but when it occurs during non-communicable disease it is often damaging and contributes to an acceleration of pathology and a worse outcome. An important inflammatory process in disease is the activation of a multi-molecular complex called the NLRP3 inflammasome (Fig. 1)1. The NLRP3 inflammasome consists of a pattern recognition receptor (PRR), which in this case is NLRP3 (NOD-like receptor (NLR) family, pyrin domain–containing protein 3 (NLRP3)), an adaptor protein called ASC (apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (CARD)), and pro-caspase-12. Described mainly in cells of haematopoietic lineage NLRP3 requires priming by pathogen associated molecular patterns (PAMPs) and subsequently becomes activated by a further PAMP, or by damage associated molecular patterns (DAMPs) causing a disruption to cellular homeostasis1. A commonly described DAMP activating NLRP3 is high levels of extracellular ATP which is sensed by the cell surface P2X7 receptor3. Activation of P2X7 induces efflux of K+ causing the association of the protein NEK7 (never in mitosis A-related kinase 7) to NLRP3 facilitating its activation4. Active NLRP3 then nucleates the oligomerisation of ASC molecules into inflammasome ‘specks’ which provide the platform for the proximity-induced auto-activation of caspase-15. Caspase-1 then cleaves the cytokine precursor molecules pro-IL-1β and pro-IL-18 to active molecules which are then secreted through an unconventional secretory route involving gasdermin D pores to the extracellular space where they drive inflammation6,7,8. Once formed the ASC specks can also be released and are stable in the extracellular environment where they further propagate inflammatory processes9,10.

Inflammasome pathway and inhibitors. The action of LPS on TLR4 induces the translocation of NFκB to the nucleus and triggers the transcription of pro-IL-1β and NLRP3. A second signal (e.g. ATP acting at P2X7), causes NLRP3, ASC and pro-caspase-1 to oligomerize and form an inflammasome speck, which permits the recruitment and activation of caspase-1 and the subsequent cleavage of pro-IL-1β into its active form which is then released. The inhibitors were added directly before the second signal, and were characterised as P2X7 receptor inhibitors, a caspase-1 inhibitor, or the NLRP3 inhibiting diarylsulfonylurea and NBC series inhibitors. The outline of the cell is courtesy of Servier Medical Art.
The NLRP3 inflammasome and IL-1β are implicated in diverse and major diseases including Alzheimer’s disease11,12, diabetes13, cardiovascular disease14, and many others. The importance of IL-1β to disease was highlighted following the recent publication of the CANTOS trial, where patients with a history of myocardial infarction were treated with canakinumab, a monoclonal antibody targeting IL-1β15. In the CANTOS trial it was found that canakinumab treatment reduced the rate of recurrent cardiovascular events, and cancer mortality, in addition to many other clinical outcomes15. However, biologicals such as canakinumab may not be suitable for the treatment of diseases where penetrance across the blood brain barrier is important, and so a small molecule inhibitor of NLRP3/IL-1 is desirable.
A number of small molecule inhibitors for the P2X7-NLRP3-Caspase-1 axis have been described16. The aim of this research was to take a selection of what we considered to be the most promising lead compounds from the literature. We focussed our study on known and potent molecules for defined points in the pathway which included antagonists of the P2X7 receptor (CE-224,53517, AZD905618, and two 5,6-dihydro-[1,2,4]triazolo[4,3-a]-pyrazine P2X7 antagonists (compounds 25 and 26 from19), the diarylsulfonylurea series (glyburide through to the cytokine release inhibiting drugs (CRIDs), including MCC95020,21,22), the caspase-1 inhibitor belnacasan (VX-765)23, and compare these to several analogues of the recently described Novel Boron Compound (NBC) inflammasome inhibitor series of boron-containing inhibitors24 (Fig. 1). This selection of molecules is by no means comprehensive and it is important to acknowledge the recent development of additional NLRP3 inhibitors not tested here such as CY-0925, and OLT117726. All molecules were tested in pre- and post-differentiated human macrophage THP1 cells using ASC speck formation and IL-1β release as endpoints, and in primary cultures of mouse microglia using IL-1β release as an endpoint. Thus we are now able to provide quantitative and comparable data for some of the most potent P2X7-NLRP3-caspase-1 inhibitors available.
Results
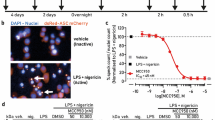
The structures of the molecules tested in the assays reported below are shown in Table 1. Human THP1 cells stably expressing ASC-Cerulean9 were either left undifferentiated or differentiated with phorbol 12-myristate 13-acetate (PMA, 0.5 µM, 3 h) before priming with bacterial endotoxin (LPS, 1 µg/ml overnight). Cells were then treated with the pan-caspase inhibitor ZVAD (50 µM, 30 min) to prevent pyroptosis and 10 point concentration response curves for each inhibitor were generated in triplicate measuring ASC speck formation in response to the K+ ionophore nigericin (10 µM, 1 h) using the IN Cell Analyzer 2000 imaging system (Fig. 2, and see methods). Under the current experimental conditions the THP1 cells failed to respond to ATP (up to 10 mM), so P2X7 inhibition was not evaluated in these cells. The results of the screen can be seen in Table 2. From the diarylsulfonylurea inflammasome inhibitor series (including glyburide, CP-412,245, CP-424,17420, CRID1, CRID221, and MCC95022), the most potent inhibitor of ASC speck formation was MCC950 with an IC50 of 3 nM in undifferentiated THP1 cells (Table 2). From the NBC inflammasome inhibitor series24 the best inhibitor was NBC19 with an IC50 of 60 nM based on speck formation in differentiated THP1 cells (Table 2). The caspase-1 inhibitor VX-765 had no effect, as expected, as the formation of the ASC speck does not require caspase-1 (Table 2). The above experiment was repeated in differentiated THP1 cells except without ZVAD and with IL-1β release as the endpoint (Table 3). Under these conditions the best inhibitor from the diarylsulfonylureas was still MCC950 with an IC50 of 4 nM (Table 3). NBC19 was still the best of the NBC series with an IC50 of 80 nM for inhibiting nigericin induced IL-1β release, and in this assay VX-765 inhibited IL-1β release with an IC50 of 10 nM (Table 3).

Compounds targeting NLRP3 but not downstream caspase-1 inhibit nigericin-induced formation of ASC specks in THP1-ASC-Cerulean macrophages. THP1-ASC-Cerulean macrophages were seeded in 96 well plate format, differentiated with PMA (0.5 µM, 3 h), pre-treated with compound for 30 min (VX-765 (a), MCC950 (b) and NBC19 (c)) before being stimulated with nigericin (10 µM, 1 h), fixed and imaged on GE InCell2000. Specks can this way be visualized and counted. Compounds targeting the NLRP3 inflammasome assembly induced a dose-dependent reduction in the number of specks. Dose response curves (d) showing the number of formed specks and the cell death for each compound. Scale bar depicts 20 µm. Respective concentration of compound and DMSO controls are indicated in the images. Data represent 3 separate cultures.
We next tested the molecules in cells relevant to CNS disease. Cultured mouse neonatal microglia were prepared as described previously27 where inflammasome responses are known to be microglia dependent27,28. Cells were primed with LPS (1 μg/ml, 3 h), then incubated with the inhibitors using a 10-point concentration response curve, before incubation with either nigericin (10 μM, 1 h), or ATP (5 mM, 1 h), in a minimum of four separate experiments. IL-1β release was measured as the endpoint. The best inhibitor from the diarylsulfonylurea series was again MCC950 with an IC50 of 60 nM against ATP-induced IL-1β release (Table 3). The P2X7 antagonists had no effect against nigericin-induced IL-1β release but were effective against ATP with AZD9056 and compound 26 (from19) both inhibiting ATP-induced IL-1β release with an IC50 of 30 nM (Table 3). NBC19 was again the most effective of the NBC series with an IC50 of 850 nM against ATP-induced IL-1β release (Table 3). VX-765, the caspase-1 inhibitor, inhibited ATP-induced IL-1β release with an IC50 of 50 nM (Table 3). There is debate in the literature as to whether responses are conserved between cultured neonatal microglia and adult microglia29,30. Thus we isolated primary adult mouse microglia as previously described31, and treated them as above (e.g. LPS, 1 μg/ml, 3 h followed by 15 min of inhibitor, followed by 5 mM ATP for 1 h). At this stage we selected the 4 best compounds across the respective classes (i.e. the P2X7 inhibitor compound 26 from19, MCC950, NBC19, and VX-765; dose responses to ATP and nigericin shown in Fig. 3a and b respectively). ATP-induced NLRP3 inflammasome activation and IL-1β secretion was conserved in isolated adult microglia and the 4 inhibitors (all tested at 10 μM, n = 10) effectively inhibited IL-1β release (Fig. 3c).

Inhibition of IL-1β secretion in neonatal and adult microglia. 50 × 104 cells were treated with LPS (1 μg/ml, 3 h), then with the selected inhibitors (VX-765, P2X7R inhibitor (compound 26 from reference19), NBC19 and MCC950, 10 μM for 15 min), and then activated with ATP (a, 5 mM, 1 h) or nigericin (b, 10 μM, 1 h). Graphs are presented showing cell death curves (in grey) and the inhibition of IL-1β release (in black) from neonatal cultures of microglia (a and b). The four inhibitors tested significantly reduced the secretion of IL-1β both in neonatal microglia and adult microglia (c). Statistical significance (vs. LPS + ATP): *p < 0.05; **p < 0.01; nd: not-detectable. Scale bar = 200 µm.
Discussion
Given the established role for IL-1 in human disease32, and preclinical evidence showing the involvement of NLRP3 in many major diseases including Alzheimer’s disease11,12,33, diabetes34, and cardiovascular disease14, the NLRP3 inflammasome is emerging as a potential drug target. Here we have characterised and compared a number of compounds known to be inhibitory across the various points in the NLRP3 pathway (Fig. 1). By testing the compounds side by side in validated assays we have been able to show that there are potent inhibitors for a number of steps of the NLRP3 pathway. The P2X7 inhibitor compound 26 is of interest for use in CNS models of disease as it is known to have high permeability across the blood brain barrier19, and we show here that it is very effective at inhibiting inflammasome activation and IL-1β release against ATP (Tables 2 and 3). Compound 26 did not inhibit nigericin induced NLRP3-dependent IL-1β release (Tables 2 and 3) confirming it as a potent and selective inhibitor of the P2X7-NLRP3 axis. As reported previously, MCC950 is a potent and selective inhibitor of NLRP3 inflammasomes in vitro and in vivo22 (Tables 2 and 3). Caspase-1 activation is an outcome of inflammasome activation and so we predicted the caspase-1 inhibitor VX-765 would be effective at inhibiting IL-1β release but not ASC speck formation which was confirmed (Tables 2 and 3). That VX-765 did not inhibit ASC speck formation confirmed that it was targeting caspase-1 specifically downstream of inflammasome activation (Table 2, Fig. 2). The comparison between the diarylsulfonylurea series20,21,22, and our NBC series24, showed that the best diarylsulfonylureas were in general more potent, and the most potent of them, MCC950, was the most active compound tested (Tables 2 and 3). Our recent work described NBC6 (with an IC50 of 570 nM) as an effective inhibitor of the NLRP3 inflammasome in vitro and in vivo24. We report here that NBC19 has improved activity, with a significantly improved IC50 of 60 nM in the ASC inflammasome speck assay in differentiated THP1 cells (Table 2). Thus we have characterised a ‘tool kit’ for dissecting inflammasome dependent responses identifying potent and well characterised reagents.
Here we established 2 protocols for assaying the effects of the inhibitors. The assay developed using the THP1-Cerulean cells9, was particularly sensitive, consistently yielding lower IC50 values than the primary glial cultures (Tables 2 and 3). Given that the IC50 established from the differentiated THP1 cells for IL-1β release more or less mirrored the IC50 obtained in the ASC speck assay, we think that the difference between the THP1 cells and the glia likely reflects the inherent variability between primary cultures when compared to using a pure clonal cell population. We also established that inflammasome responses are robust and comparable in acutely isolated primary microglia from the adult brain (Fig. 3c), giving confidence to the data generated using neonatal microglia.
It is important to note, that while our research has focussed on targeting the NLRP3 inflammasome in innate immune cells, NLRP3 is also important in non-immune cell function. For example, the NLRP3 inflammasome is present in epithelial cells in many tissues and is known to be involved in many physiological processes35. There is evidence that epithelial cell NLRP3 also regulates caspase-1-dependent release of pro-inflammatory cytokines such as IL-1β (e.g.36), but it is also reported to have other functions such as regulating the activation of sterol regulatory element binding proteins (SREBPs) which repair the plasma membrane of epithelial cells in response to pore forming bacterial toxins37. Thus the ‘toolkit’ described here will also be of value to scientists studying NLRP3 function in non-immune cells such as epithelial cells.
In summary, the data provided here give comparable quantitative data on inhibitors of the NLRP3 inflammasome pathway and identify their selectivity across the particular points of intervention, identifying them as a valuable tool kit for interrogating inflammasome dependent responses in cell based models.
Materials and Methods
Inhibitors
The NBC molecules were synthesised and prepared at the University of Manchester as described previously24. MCC950 (Sigma-Aldrich, Dorset, UK) and VX-765 (Selleckchem, Munich, Germany) were purchased. All other compounds in the P2X7 and diarylsulfonylurea series were synthesised following the relevant published literature either in house at LifeArc or at GVK Biosciences, Bengaluru, India.
THP1 culture and differentiation
ASC-Cerulean expressing THP1 cells were grown in RPMI, 10% FBS and 1% Pen/Strep. Cells were differentiated using 0.5 µM PMA for 3 h and used for assays the next day.
NLRP3 inflammasome activation in THP1 cells
ASC-Cerulean expressing THP1 cells were pre-incubated with compound (1% final DMSO) for 30 min before being stimulated with 10 µM nigericin for 1 h. Pre-differentiated cells were treated with the pan caspase inhibitor Z-VAD-FMK (Promega, Southampton, UK; 50 µM, 30 min) during compound incubation. Supernatants were collected for quantification of IL-1β using mesoscale Tissue Culture Kit (K151AGB) and cells were fixed for the speck assay.
Speck assay and analysis
ASC-Cerulean expressing THP1 cells were fixed with 4% formaldehyde for 15 min at room temperature, washed with PBS and stained with HCS Red (Invitrogen, Loughborough, UK) to visualize nuclei. Images were acquired on the GE IN Cell 2000 and analysed in Workstation to count both nuclei and cerulean-labelled ASC specks.
Preparation of neonatal glial cultures
Mice (C57BL/6 strain) were maintained under standard laboratory conditions (20 ± 2 °C, 12-h light cycle, humidity of 40–50%, 12 h light cycle, ad libitum access to water and standard rodent chow). All procedures were performed by appropriate personal and under project licences, in accordance with the Home Office (Animals) Scientific Procedures Act (1986) and approved by the Home Office and the local Animal Ethical Review Group, University of Manchester. Each litter was considered as an n number, and a minimum of 3 litters was used to test each compound.
Brains were harvested from 3–4 days old mice under aseptic conditions, and olfactory bulbs, cerebellum, and meningeal layers, were gently removed. Brain tissue was mechanically digested, centrifuged, and cells resuspended in media and seeded on cell culture flasks. Cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Sigma-Aldrich), 10% fetal bovine serum (FBS, Life Technologies, Warrington, UK), 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma-Aldrich). Media was changed every 4–5 days until cells reached 80% confluence (around day 10–13 in vitro (div)). Cells were then trypsinized (0.5% Trypsin EDTA, 2 min at 37 °C, Sigma-Aldrich) and gently scraped, counted and seeded on plates (1.7 × 105 cells/ml). After 2–3 days, cells were ready for further experiments.
Preparation of adult microglia
Adult C57BL/6 mice were perfused with ice-cold Hank’s Balanced Salts Solution (HBSS) and brains kept in cold HBSS. After removal of cerebellum and meningeal layers, brains were diced, and enzymatically processed using as per manufacturer’s instructions using a MACS Neural Tissue Dissociation Kit (P) (Miltenyi Biotech, Bisley, UK). Brains were then mechanically homogenised using a Dounce homogeniser, and myelin removed from the resulting cell suspension using a one-step 30% Percoll gradient31. Microglia were then isolated using magnetic CD11b + beads (Milteny Biotech), and seeded on poly-L-lysine coated plates (~1.7 × 105 cells/ml), and maintained with DMEM (with 10% fetal bovine serum and 1% penicillin streptomycin) supplemented with 10 ng/ml of recombinant mouse M-CSF (R&D systems, Abingdon UK), and 50 ng/ml of recombinant human TGF-β. Media was changed at 3 days, and cells treated after 7 days in culture.
Inflammasome activation assays in glia
Fresh media (DMEM, 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin) was added before starting the treatments. Cells were treated with gel filtration chromatography purified LPS (1 µg/ml, 3 h, E.coli O26:B6, L2654, Sigma-Aldrich) and then the media was changed to serum free media before the addition of drugs that were dissolved in DMSO (except MCC950, in PBS), and were added as a 10 point dose response from 0.01–100 µM (compounds shown in Table 1). After 15 min incubation with the drugs (or vehicle), the NLRP3 inflammasome was activated as follows by adding ATP (5 mM) or nigericin (10 µM) for 1 h. Supernatants were collected and used for further analysis of cell death and IL-1β release.
Lactate dehydrogenase measurement of cell death
Lactate dehydrogenase (LDH) release was used as a measure of cell death using a CytoTox 96® Non-Radioactive Cytotoxicity Assay (Promega). Following manufacturer’s instructions, absorbance at 490 nm was measured and converted into cell death values.
IL-1β measurements
THP1 cells
Secretion of IL-1β was quantified using mesoscale Tissue Culture Kit (K151AGB) following the manufacturer’s instruction.
Microglial cells
Secretion of IL-1β was quantified by ELISA using DuoSet® kits (R&D Systems) following the manufacturer’s instructions.
Statistical analysis
THP1 cells
For both readouts, speck assay and IL-1β quantification, half maximal inhibitory concentration (IC50) of drugs was determined by fitting the data to a four parameter logistic equation (Nonlinear Regression, Sigmoidal, 4PL) in GraphPad Prism version 7.03. Mean and SEM values are plotted from at least 3 experiments.
Glia
For each assay IL-1β levels were calculated against a four-parameter logistic (4-PL) curve fit, using GraphPad Prism version 7.00 for Windows, GraphPad Software (USA). All values are expressed as a percentage of vehicle conditions or as mean ± standard error of the mean (SEM), and a minimum of 3 litters (n = 3) was used for each tested compound. The half-maximal inhibitory concentration (IC50, μM) for each drug was determined by fitting the data to a four-parameter logistic equation using GraphPad Prism. One way ANOVA was performed on IL-1β values for the adult and neonatal microglia experiments (comparing vs. LPS + stimulus). Holm-Sidak post hoc tests were performed if statistical significance (p value < 0.05) was achieved, and indicated in the graphs as follows: *p < 0.05; **p < 0.01; ***p < 0.001.
Data availability
The data that support the findings of this study are available from the corresponding author on request.
References
Liston, A. & Masters, S. L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nature reviews. Immunology 17, 208–214, https://doi.org/10.1038/nri.2016.151 (2017).
Broz, P. & Dixit, V. M. Inflammasomes: mechanism of assembly, regulation and signalling. Nature reviews. Immunology 16, 407–420, https://doi.org/10.1038/nri.2016.58 (2016).
Mariathasan, S. et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232, https://doi.org/10.1038/nature04515 (2006).
He, Y., Zeng, M. Y., Yang, D., Motro, B. & Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357, https://doi.org/10.1038/nature16959 (2016).
Dick, M. S., Sborgi, L., Ruhl, S., Hiller, S. & Broz, P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nature communications 7, 11929, https://doi.org/10.1038/ncomms11929 (2016).
Lopez-Castejon, G. & Brough, D. Understanding the mechanism of IL-1beta secretion. Cytokine & growth factor reviews 22, 189–195, https://doi.org/10.1016/j.cytogfr.2011.10.001 (2011).
Brough, D., Pelegrin, P. & Nickel, W. An emerging case for membrane pore formation as a common mechanism for the unconventional secretion of FGF2 and IL-1beta. Journal of cell science 130, 3197–3202, https://doi.org/10.1242/jcs.204206 (2017).
Evavold, C. L. et al. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44 e36, https://doi.org/10.1016/j.immuni.2017.11.013 (2018).
Franklin, B. S. et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nature immunology 15, 727–737, https://doi.org/10.1038/ni.2913 (2014).
Baroja-Mazo, A. et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nature immunology 15, 738–748, https://doi.org/10.1038/ni.2919 (2014).
Heneka, M. T. et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678, https://doi.org/10.1038/nature11729 (2013).
Daniels, M. J. et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nature communications 7, 12504, https://doi.org/10.1038/ncomms12504 (2016).
Masters, S. L. et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nature immunology 11, 897–904, https://doi.org/10.1038/ni.1935 (2010).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361, https://doi.org/10.1038/nature08938 (2010).
Ridker, P. M. et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. The New England journal of medicine. https://doi.org/10.1056/NEJMoa1707914 (2017).
Baldwin, A. G., Brough, D. & Freeman, S. Inhibiting the Inflammasome: A Chemical Perspective. Journal of medicinal chemistry 59, 1691–1710, https://doi.org/10.1021/acs.jmedchem.5b01091 (2016).
Stock, T. C. et al. Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. The Journal of rheumatology 39, 720–727, https://doi.org/10.3899/jrheum.110874 (2012).
Keystone, E. C. et al. Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Annals of the rheumatic diseases 71, 1630–1635, https://doi.org/10.1136/annrheumdis-2011-143578 (2012).
Chrovian, C. C. et al. Novel Phenyl-Substituted 5,6-Dihydro-[1,2,4]triazolo[4,3-a]pyrazine P2X7 Antagonists with Robust Target Engagement in Rat Brain. ACS chemical neuroscience 7, 490–497, https://doi.org/10.1021/acschemneuro.5b00303 (2016).
Perregaux, D. G. et al. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. The Journal of pharmacology and experimental therapeutics 299, 187–197 (2001).
Laliberte, R. E. et al. Glutathione s-transferase omega 1-1 is a target of cytokine release inhibitory drugs and may be responsible for their effect on interleukin-1beta posttranslational processing. The Journal of biological chemistry 278, 16567–16578, https://doi.org/10.1074/jbc.M211596200 (2003).
Coll, R. C. et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature medicine 21, 248–255, https://doi.org/10.1038/nm.3806 (2015).
Wannamaker, W. et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoy l)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. The Journal of pharmacology and experimental therapeutics 321, 509–516, https://doi.org/10.1124/jpet.106.111344 (2007).
Baldwin, A. G. et al. Boron-Based Inhibitors of the NLRP3 Inflammasome. Cell Chem Biol 24, 1321–1335 e1325, https://doi.org/10.1016/j.chembiol.2017.08.011 (2017).
Jiang, H. et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. The Journal of experimental medicine 214, 3219–3238, https://doi.org/10.1084/jem.20171419 (2017).
Marchetti, C. et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proceedings of the National Academy of Sciences of the United States of America 115, E1530–E1539, https://doi.org/10.1073/pnas.1716095115 (2018).
Savage, C. D., Lopez-Castejon, G., Denes, A. & Brough, D. NLRP3-Inflammasome Activating DAMPs Stimulate an Inflammatory Response in Glia in the Absence of Priming Which Contributes to Brain Inflammation afterInjury. Frontiers in immunology 3, 288, https://doi.org/10.3389/fimmu.2012.00288 (2012).
Gustin, A. et al. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PloS one 10, e0130624, https://doi.org/10.1371/journal.pone.0130624 (2015).
Schell, J. B., Crane, C. A., Smith, M. F. Jr & Roberts, M. R. Differential ex vivo nitric oxide production by acutely isolated neonatal and adult microglia. Journal of neuroimmunology 189, 75–87, https://doi.org/10.1016/j.jneuroim.2007.07.004 (2007).
Brannan, C. A. & Roberts, M. R. Resident microglia from adult mice are refractory to nitric oxide-inducing stimuli due to impaired NOS2 gene expression. Glia 48, 120–131, https://doi.org/10.1002/glia.20066 (2004).
Nikodemova, M. & Watters, J. J. Efficient isolation of live microglia with preserved phenotypes from adult mouse brain. Journal of neuroinflammation 9, 147, https://doi.org/10.1186/1742-2094-9-147 (2012).
Dinarello, C. A., Simon, A. & van der Meer, J. W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nature reviews. Drug discovery 11, 633–652, https://doi.org/10.1038/nrd3800 (2012).
Venegas, C. et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 552, 355–361, https://doi.org/10.1038/nature25158 (2017).
Hu, C. et al. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proceedings of the National Academy of Sciences of the United States of America 112, 11318–11323, https://doi.org/10.1073/pnas.1513509112 (2015).
Santana, P. T. et al. Is the inflammasome relevant for epithelial cell function? Microbes and infection/Institut Pasteur 18, 93–101, https://doi.org/10.1016/j.micinf.2015.10.007 (2016).
Khalafalla, M. G. et al. P2X7 receptor antagonism prevents IL-1beta release from salivary epithelial cells and reduces inflammation in a mouse model of autoimmune exocrinopathy. The Journal of biological chemistry 292, 16626–16637, https://doi.org/10.1074/jbc.M117.790741 (2017).
Gurcel, L., Abrami, L., Girardin, S., Tschopp, J. & van der Goot, F. G. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126, 1135–1145, https://doi.org/10.1016/j.cell.2006.07.033 (2006).
Dorokhov, V. A., Vasil’ev, L. S., Surzhikov, F. E. & Bogdanov, V. S. Chelate synthesis of 3-ethoxycarbonyl-4-hydroxy-2-trifluoromethylpyridine from ethyl acetoacetate and trifluoroacetonitrile. Russian Chemical Bulletin 44, 1283–1285 (1995).
Vasil’ev, L. S., Azarevich, O. G., Bogdanov, V. S., Bochkareva, M. N. & Dorokhov, V. A. Boron chelates with 5,5,5-trifluoro- and 5,5,5-trichloro-4-aminopent-3-en-2-ones. Bulletin of the Russian Academy of Sciences, Division of chemical science 41, 2104–2107 (1992).
Acknowledgements
Thanks to Eicke Latz (Bonn, Germany) for the ASC-Cerulean expressing THP1 cells. This work was funded by a ARUK Dementia Consortium grant (ARUK-DC2017-1), and in part by a MRC Momentum grant (MC_PC_16033).
Author information
Authors and Affiliations
Contributions
E.R.C., D.F., M.J.H., A.G.B., S.O. performed experiments and analysed and interpreted data. S.F., E.K., H.N., P.J.A., L.A.D., C.R., S.M.A., S.F., J.B., and D.B. conceived and planned the study and obtained funding. All authors contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Redondo-Castro, E., Faust, D., Fox, S. et al. Development of a characterised tool kit for the interrogation of NLRP3 inflammasome-dependent responses. Sci Rep 8, 5667 (2018). https://doi.org/10.1038/s41598-018-24029-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-24029-3
This article is cited by
-
Pulmonary toxicity assessment of polypropylene, polystyrene, and polyethylene microplastic fragments in mice
Toxicological Research (2024)
-
Pyroptosis in bone loss
Apoptosis (2023)
-
Spermidine reduces neuroinflammation and soluble amyloid beta in an Alzheimer’s disease mouse model
Journal of Neuroinflammation (2022)
-
Synthesis, biological evaluation and molecular modeling studies of novel 1,2,3-triazole-linked menadione-furan derivatives as P2X7 inhibitors
Journal of Bioenergetics and Biomembranes (2022)
-
A phenotypic high-content, high-throughput screen identifies inhibitors of NLRP3 inflammasome activation
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
