
Abstract
Female fertility is a highly regulated process involving the synchronized activities of multiple tissues. The underlying genomic regulation of the tissue synchronization is poorly understood. To understand this better we investigated the transcriptomes of the porcine ovary, endometrium, and oviduct at days 0, 3, 6, 9, 12, 15, or 18 of the oestrous cycle. We analysed the transcriptome profiles of the individual tissues and focus on the bridging genes shared by two or more tissues. The three tissue-networks were connected forming a triangular shape. We identified 65 bridging genes with a high level of connectivity to all other genes in the network. The expression levels showed negative correlations between the ovary and the other two tissues, and low correlations between endometrium and oviduct. The main functional annotations involved biosynthesis of steroid hormones, cell-to-cell adhesion, and cell apoptosis, suggesting that regulation of steroid hormone synthesis and tissue viability are major regulatory mechanisms.
Similar content being viewed by others

Introduction
Comprehensive understanding of the regulatory mechanism of the oestrous cycle in domestic animals is important for manipulating animal productivity and resolving reproductive problems such as infertility1. This is of particular interest in pigs, for which reproductive rate and litter size impact commercial value. The complicated range of hormone secretions and physiological changes that appear during the oestrous cycle are closely linked to multiple molecular interactions among a number of reproduction-related tissues, including the hypothalamus, the pituitary, the ovary, the oviduct, and the endometrium2. The oestrous cycle lasts an average of 21 days in female pigs. At the start of the oestrous cycle, levels of estrogen are highest and the release of pre-ovulatory gonadotrophin releasing hormone (GnRH) and luteinising hormone (LH) occurs as a result of the positive feedback mechanism of estrogen action, and ovulation follows. Then, levels of progesterone begin to increase after ovulation and decrease at late diestrus when the implanting conceptuses are not present in the uterus and luteolysis is induced.
The cyclic and sequential secretion of hormones from each reproductive tissue regulates morphological and functional changes in reproductive tissues to mediate the progression of the oestrous cycle3,4. The changes in hormonal milieu control ovarian activity which leads to follicular development, ovulation, luteinisation and luteolysis5. The ovarian activity governs the oviductal function for the sperm capacitation, fertilization and preimplantation embryo development, and the endometrial function regulatory for conceptus implantation and the establishment and maintenance of pregnancy6,7,8. In addition, the ovary acts on the hypothalamus and the pituitary glands at a positive and negative feedback mechanism for regulation of hormone production of those tissues. The uterine endometrium is involved in regulation of ovarian function by producing the luteolytic signal, prostaglandin F2α(PGF2α), at the end of the oestrous cycle in domestic animal species. Thus, the cyclicity of the oestrous cycle is the result of complicated but well-coordinated reciprocal interactions among reproductive tissues9,10,11. However, the regulatory mechanism of the oestrous cycle at the cellular and molecular levels and the expression and function of genes in reproductive tissues are not fully understood.
Transcriptome changes in the ovary, the endometrium, and the oviduct have been studied under different experimental conditions. The oviductal transcriptome has shown to be influenced by the ovarian hormones such as estrogen and progesterone12,13. Ovarian transcriptome analyses revealed genes that related to litter size in pigs14. In addition, transcriptomic changes in the endometrial tissue have been analysed at the time of implantation and during pregnancy15,16. In humans, many studies have focused on the changes in the transcriptomes by the presence of the embryo or due to reproductive failure in the endometrium and oviduct17,18. However, it has not been attempted to investigate the dynamic regulatory mechanism in the transcriptomes and the coordination of the transcriptome changes in multiple reproductive tissues during different phases of the reproductive cycle in any animal species.
The objective of this study was to investigate the molecular mechanisms underlying the synchronization of the ovary, endometrium and oviduct reproductive tissues in the pig. We identified the differentially expressed genes during the different phase of the oestrous cycle and among different reproductive tissues and the key genes in each network to elucidate the biological roles of these genes in the reproductive tracts. Then, we integrated the transcriptomes of the ovary, the endometrium, and the oviduct and analyse both universal and tissue-specific gene regulation throughout the oestrous cycle using RNA-Seq analysis. Dynamic molecular networks from the integrated transcriptomes were elucidated by the gene co-expression network (GCN) analysis. Here, we report that differentially expressed genes shared among the reproductive tissues in a universal and tissue-specific manner may be involved in regulating the cyclicity of the oestrous cycle in pigs.
Results
Integration of reproductive tissue transcriptomes during the oestrous cycle
We obtained reproductive tissues from the endometrium, ovary, and oviduct, and analysed the transcriptomes expressed in those tissues on days 0, 3, 6, 9, 12, 15, and 18 of the oestrous cycle (Fig. 1A). The RNA-seq data revealed clear differences among the three tissues and the transcriptomes from different time points throughout the oestrous cycle were clustered within individual tissues (Fig. 1B). To better understand the dynamic changes of each tissue-specific transcriptome throughout the oestrous cycle, we compared transcriptomes of each tissue at each time point to the transcriptome of corresponding tissue on day 0 and determined the differently expressed genes (DEGs) (Fig. 1C). Large changes in gene expression were observed between day 6 and day 9 in the endometrial and ovarian transcriptomes and between day 9 and day 12 in the endometrial and oviductal transcriptomes (DEGs > 3,000 transcripts). We also compared the number of significant DEGs among the three tissues at each time point. The Venn diagrams representing the overlap DEGs between the tissues at each time point revealed that the number of overlap DEGS increased until day 9 and then rapidly decreased at the end of the cycle indicating genomic regulation of the reproduction processes.

Overview of reproductive tissue transcriptomes by passing through a swine oestrous cycle. (A) Transcriptomes in three female reproductive tissues (endometrium, ovary and oviduct) for 7 dates (day 0, D00; day 3, D03; day 6, D06; day 9, D09; day 12, D12; day 15, D15 and day 18, D18) on the phases of the oestrous cycle. (B) A principal component analysis revealed far distances between different tissue groups and clusters among the seven time points transcriptomes based on the oestrous cycle within tissue groups. (C) Dynamic view of DEGs based on the start of oestrous cycle (D00). The x-y axes of scatter plots were scaled by log10 and log2 fold changes. Significant DEGs (FDR < 0.05) in the scatter plots were coloured and counted by red dots and a number. Venn diagrams with scaled bars revealed numbers of integrated DEGs by each of time points and tissues. The numbers over 100 in the tissue specific Venn diagrams were indicated by red colour.
GCN of the porcine oestrous cycle
Next, we integrated the entire transcriptomes from three reproductive tissues at seven time points during the oestrous cycle using GCN analysis to study DEG shared by two or three tissues at the same time points. A total of 904 genes were employed in the PCIT algorithm that combines the concept partial correlation coefficient with information theory to estimate significant connections between genes. A GCN was constructed with 622 nodes (genes) and the nodes were connected by 9,639 edges (Fig. 2). The network was comprised of three clearly clustered tissue-specific sub-networks representing each tissue with a small number of genes connecting the sub-networks. The sub-networks also revealed different expression patterns based on k-mean expression clustering analysis (Supplementary data 2). We observed that an endometrium-specific sub-network (red) showed changes in gene expression between different time points of the oestrous cycle, while the other tissues did not show any changes in gene expression. Moreover, the two different expression clusters in the endometrium represented a sub-network of upregulated genes and a sub-network of downregulated genes, which appeared between days 6 and 12. In the ovary-specific sub-network (yellow), the expression changes were noticeable between days 3 and 12, with both up- and downregulations represented as clusters 3 and 4, respectively. The oviduct-specific sub-network had one expression cluster with downregulated genes, whereas the other two tissues showed a slightly upregulated expression pattern.

GCN of the porcine oestrous cycle. The network shows three substantial core groups (represented by different node colours), which were clearly separated by each reproductive tissue: endometrium (red), ovary (yellow), and oviduct (blue). The PCIT algorithm generated significant connections and a nominal threshold (r2 > 0.90) was applied to the network analysis. The overall GCN contained 622 genes connected by 9,639 edges. Node size and transparency were mapped to the highest log2 FC at each time point. Edge transparency was mapped in-between nodes. Gene clustering analysis by the k-means clustering algorithm in the MultiExperiment Viewer software revealed five distinct expression profiles across the oestrous phases (left to right) and tissues (same colour with node). The levels of expression were presented as 3 and −3 log2 FC. The total number of genes and the percentages in the brackets are located at the top left corner in the cluster.
The DAVID enrichment analysis for KEGG pathway terms showed significantly enriched pathways (Fig. 3). There was an enrichment of genes in the network that are a part of several biological pathways involved in the female reproductive system, including haemostasis, signalling in immune system, metabolism of lipids and lipoproteins, extracellular matrix-receptor interaction, and terpenoid backbone biosynthesis19,20. In addition, several pathways related to metabolism of prostaglandins and steroid hormones were included. These pathways are important components of the immune protection of the female reproductive tract and affect fertilization and pregnancy success.

Enrichment analyses of biological meaning in the network. Enrichment analyses of the pathways from all databases in DAVID. The significant pathways in the chart passed a cut-off (−log10 P-value > 1.0).
Tissue-specific networks and functional annotation in the oestrous cycle
Based on the presence of tissue-specific sub-networks, we integrated the biological differences and similarities among three reproductive tissues throughout the oestrous cycle. First we investigated the tissue-specific sub-networks separately. Genes were excluded from the independent network if they were not directly connected within the main sub-networks. We provided list of genes involved in each of tissue-specific sub-networks (Supplementary data 3). The independent networks represent the connections between genes that showed the largest FC values in the different phases of the oestrous cycle (Fig. 4A–C).

Tissue-specific sub-networks. Sub-networks were reconstructed from the original network in Fig. 2 and enrichment analyses for the genes in the subnetworks were performed. Endometrium (A,D, and G), ovary (B,E, and H), and oviduct (C,F, and I). (A–C) Differently coloured nodes represent the expression values among the oestrous cycle phases and their values are shown in brackets. The number of nodes for each colour is shown in brackets. (D–F) DAVID-enriched pathways for the tissue-specific subnetworks after applying the cut-off (−log10 P-value > 1.0 and the number of responsible genes > 3). (G–I) Gene Ontology treemaps regarding biological process terms for the three reproductive tissues. Enriched GO terms are indicated by bold letters.
The endometrium-specific network consisted of 216 DEGs with 1,501 significant connections (r2 > 90). In the endometrium-specific network, day 9 had the highest number of genes with the maximum FC level (D09, n = 88), followed by day 6 (D06, n = 60) and day 12. There were genes that showed the maximum FC level from day 3 until the end of the oestrous cycle and there were only a small number of genes showing the maximum FC level on day 18 (D12, n = 50), as shown in Fig. 4A. The ovary-specific network was the largest of the three tissues, consisting of 324 genes with 7,794 connections (Fig. 4B). In the ovary-specific network, day 6 had the highest number of genes with the maximum FC level (n = 100), followed by day 9. Day 12 showed a high number of nodes, while day 15 and day 18 showed low FC levels. The oviduct-specific network was small compared with the other two networks, consisting of 69 genes with 264 connections (Fig. 4C). In the oviduct-specific network, day 3 and day 18 showed no genes with the maximum FC level of the same tissue.
In summary, the highest number of significant genes was found in the ovary, the lowest number of significant was found in the oviduct (Fig. 4). In addition, we observed different levels of expression between oestrous phases based on the number of genes with maximum FC values through the oestrous phases and three tissues. Peak gene expression was observed between days 6 and 9, whereas the end of the cycle, between days 15 and 18, revealed basal gene expression levels.
Significant pathway terms from the Kyoto Encyclopaedia of Genes and Genomes (KEGG) database were enriched in the list of genes from each of the tissue-specific networks (Fig. 4D–F). The genes with the highest differential expression in the endometrium-specific network showed significant enrichment for eicosanoid metabolism (Fig. 4D), which is related to changes in prostaglandin and inflammation in blood vessels21. The genes with the highest differential expression in the ovary-specific network exhibited enrichment in terms related to steroid hormone synthesis (Fig. 4E). Moreover, the ECM-receptor interaction was robustly pronounced under the luteinising hormone-mediated activation in ovarian cancer22. In the chart for enriched pathways of the oviduct-specific network showing the genes with the highest differential expression, the ECM-receptor interaction was also significantly enriched (Fig. 4F). The focal adhesion pathway, which has potential roles in embryo transit and placentation, has also been reported18,23. The illustrated treemaps displayed significantly enriched GO terms in biological processes (Fig. 4G–I). The highest enrichments for each tissue were: The endometrium-specific genes were broadly associated with GO terms for responses to chemicals and the regulation of multicellular organismal processes (Fig. 4G), whereas the genes in the ovary-specific network were significantly enriched with GO terms for regulation of ion transport and very long-chain fatty acid metabolism (Fig. 4H). The genes in the oviduct-specific network were significantly enriched with GO terms for collagen biosynthesis and response to chemical (Fig. 4I).
Genes involved in bridging between the tissue-specific sub-networks
A network for the entire integrated transcriptome consisted of three robust tissue-specific sub-networks, as shown in Fig. 2. Interestingly, the three sub-networks were connected with a small number of genes forming a triangular shape. Genes that connected at least two sub-networks were designated as bridging genes. In total, there were 65 bridging genes (Supplementary data 3) that had a high level of connectivity (n = 1,454) to all other genes in the network (Fig. 5A). The absolute FC expression levels of genes during the oestrous cycle were highest between day 6 and day 12 in each reproductive tissue (Fig. 5B). As shown in the heat map of the correlation matrix (Fig. 5C), there were many negative correlations between the expression levels in ovary and the other two tissues, whereas there were low correlations between the expression levels in endometrium and oviduct. Moreover, the correlation matrix revealed strong positive correlations in gene expression between day 6 and day 12 in each tissue. Days 3, 15, and 18, however, had relatively low correlations between each other.

Embedded core network by gene bridging between tissues. (A) When a gene (node) has connections (edges) with at least two different tissues (colours), it is designated as a bridging gene. Genes were condensed into a core network, which had a high number of connections with the genes of subnetworks. (B) Scattered box plots representing waves of expression in genes of the core network during the oestrous cycle. Black dots in the plots have absolute log2 FC greater than 3.0, while all other dots are grey. The colours of box plots were matched with each of the colours of the oestrous phases in Fig. 4. (C) Correlation matrix of the core-network genes. A heat map representing the correlations between genes in different oestrous phases and tissues. The scale of correlation coefficients is coloured with red, white, and blue, representing 1, 0, and −1, respectively.
The GO enrichment analysis revealed specific clustering for functional annotations of genes. The Table 1 results represented the significantly clustered GO terms such as steroid biosynthetic process and positive regulation of apoptosis that involve in the female reproductive system24. The first ranked GO cluster involved biosynthesis of steroid hormones and the following clusters involved cell-to-cell adhesion and cell apoptosis. The GO terms that we identified in the top ranked clusters of reproductive tissues commonly participate in steroidogenesis across the oestrous cycle and possibly regulate reproductive hormone signalling.
qPCR validations for specific target genes
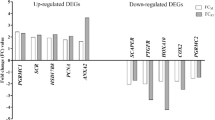
To technically validate genes found in tissue-specific networks, we performed qPCR analysis and compared the expression of those genes determined by qPCR to that by RNA-seq. We have chosen 22 genes included in biological pathways of networks specific for the endometrium, ovary, and oviduct. HSD11B1, FXYD4, PTGER2, PTGS2, CYP2J2, SGPP1, and SGPP2 were selected for biological pathways in the endometrium-specific network (aldosterone-regulated sodium reabsorption, eicosanoid metabolism, and sphingolipid metabolism), TLR2, TLR4, TLR9 AKR1C1, CYP7A1, CYP17A1, CYP19A1, ACAT2, HMGCR, HMGCS1, MVK for biological pathways in the ovary-specific network (dendritic cells in regulating Th1 and Th2 development, steroid biosynthesis, and terpenoid backbone biosynthesis), and COL1A1, COL3A1, IGF1, and ACTC1 for biological pathways in the ovary-specific network (focal adhesion and hypertrophic cardiomyopathy).
To examine global gene expression during the oestrous cycle between the two experiments, a comparative heatmap was visualized using quantile-normalized values (Fig. 6A). Most gene expression patterns were observed to be analogous between qPCR and RNA-seq throughout the oestrous cycle. To quantitatively assess this observation, the Pearson’s correlation coefficients were calculated using the log-fold change ratio comparison between the two experiments and found a significant correlation in total (r2 = 0.865 and Fig. 6B).

Comparisons of target gene expression by qPCR for technical validation. (A) Heatmap representing log2 FC expression values from both qPCR and RNA-seq. The gradient ranges from 10 (red) to −10 (blue). (B) Regression plot displaying the direct correlations between the two values for each gene. The seven different time periods of the oestrous cycle are not shown in the plot.
Localization of genes involved in steroid biosynthesis and eicosanoid metabolism in the reproductive tissues
To understand the cellular localization of the expression of genes found in GCN analysis, we performed in situ hybridization analysis. We have chosen CYP7A1, CYP17A1, and CYP19A1, genes included in a biological process, steroid biosynthesis, found in the ovary-specific network, and PTGS2 and PTGER2, genes included in a biological process, eicosanoid metabolism, found in the endometrium-specific network. As shown in Fig. 7A, the expression of CYP7A1 and CYP19A1 mRNAs was detected in the ovarian follicles with strong signal intensity in granulosa cells (GCs), while the expression of CYP17A mRNA was detected mainly in ovarian thecal cells (TCs). The expression of endometrial genes, PTGS2 and PTGER2, was localized to endometrial epithelial and stromal cells with strong signal intensity at late diestrus stage of the oestrous cycle (Fig. 7B).

In situ hybridization analysis of CYP7A1, CYP17A1 and CYP19A1 mRNAs in the ovary (A) and PTGS2 and PTGER2 mRNAs in the uterine endometrium (B) during the oestrous cycle in pigs. Representative uterine sections from indicated day of the oestrous cycle hybridized with DIG-labeled sense cDNA probes (Sense) as a negative control are shown for in situ hybridization analysis. D, Day; GC, Granulsa cell; TC, Theca cell; LE, luminal epithelium; GE, glandular epithelium; St, stroma. Scale bar = 100 μm.
Discussion
Differentially expressed genes among tissues and time of oestrus cycle
During the oestrous cycle, the female reproductive system undergoes physiological, morphological, and functional changes in multiple tissues. Hence, understanding the regulatory mechanism of the function of the reproductive tissues is essential for the control of animal productivity and diseases. In this study, we investigated the molecular mechanisms of the oestrous cycle in the female reproductive tissues in pigs. We analysed transcriptomes of the female reproductive tissues of ovary, oviduct and endometrium by comparing and integrating the three transcriptomes throughout the oestrous cycle. According to the PCA plot using the transcriptomes expressed at different time of the oestrous cycle and in different reproductive tissues, a well-defined trajectory of transcriptomes was observed depending on the tissue type. Moreover, the transcriptomes tended to cluster among the days 6, 9, and 12, and among the days 0, 15, and 18 of the oestrous cycle. When the transcriptomes were flared out by DEG values based on day 0, the similarities between tissues could be more clearly displayed. The number of significant DEGs increased until approximately day 9 and then decreased to a steady state. The drift curves of DEGs resemble the curve for progesterone (P4) secretion and can be related to the secretion of prostaglandin (PGF2α), which causes a drop in P4 secretion when a female pig undergoes luteolysis25,26. However, it was difficult to find similarities between the drift curves for DEGs and oestrogen (E2) or gonadotropins (LH and FSH).
Dynamic changes during the oestrus cycle within each tissue
According to list of the significant DEGs along the oestrus cycle of the three tissues, we noted the numbers of common and unique DEGs among the three tissues. Upon the release of GnRH and gonadotropins from the hypothalamic–pituitary axis, the oestrous cycle enters the follicular phase which leads to ovulation and the progression to the luteal phase in the ovary27. In response to progesterone from the corpus luteum (CL) in the luteal phase, the oviduct and endometrium prepare for pre-implantation embryo development and the establishment for pregnancy, whereas the endometrium initiates the luteolytic process when pregnancy is not established at the end of the luteal phase. Because the ovary, endometrium and oviduct intercommunicate and play major roles in the maintenance of the oestrous cycle, DEGs that are common among the reproductive tissues might be involved in the same biological processes for the oestrous cyclicity in all investigated tissues, while the DEGs that are unique to each of the tissue are likely to be involved in functions specific to the particular reproductive tissue.
The weighted GCN has advantages for representing system-level properties and for identifying specific molecular mechanisms28,29,30. We showed that several integrated transcriptomes can be illustrated at once by GCN analysis using the PCIT algorithm31. Only stringent DEGs (absolute FC > 3.0 and FDR < 0.05) were employed for the GCN analysis. The delta-winged network revealed three sub-networks corresponding to each of the reproductive tissues; meanwhile, the sub-networks were connected to each other with a small number of bridging genes. As revealed in the transcriptome integrations and expression pattern clusters in the constructed network, our data clearly showed the dynamic changes along the oestrous cycle in each reproductive tissue. Moreover, the significant nodes in the network displayed tissue-specific sub-networks and the genes were only highly differentially expressed within the tissue-specific sub-networks. This may suggest that the morphological changes during the oestrus cycle within each tissue individually are regulated via differential gene expression. Furthermore, this regulation within each tissue may be related to smaller differential gene expressions regulating tissue synchronization during the oestrus cycle. Dynamic changes in DEGs or significant gene subsets may represent general biological signals. Therefore, the constructed networks were used to investigate the common or specific reproductive mechanisms in the oestrous cycle can reveal specific changes in gene expression.
The importance of regulation of the steroid hormone and lipid metabolisms
Since the dynamic changes in DEGs of significant gene subsets specify the general biological actions, we constructed gene networks to investigate the common or specific functions of the reproductive tissues during the oestrous cycle. The functional annotation enrichment analysis for KEGG pathways in the constructed gene network revealed many significantly enriched terms. The enriched terms in the ovary-specific sub-network included terpenoid backbone biosynthesis, steroid biosynthesis, sterol metabolism, cholesterol transport, and lipid metabolism, which are related to steroid hormone biosynthesis and lipid metabolism. In the ovary, steroidogenesis is one of the major activities for sexual and reproductive functions32. In response to gonadotrophins, the ovary produces the major steroid hormones, estrogens and progesterone, which play important roles in reproductive tissues as well as a variety of other tissues. It is known that reproduction is directly coupled with lipid metabolism, which is also affected by steroid hormones33,34. Furthermore, it has been shown that the DEGs in the ovarian transcriptomes of pigs differing in litter size are involved in steroid biosynthesis, steroidogenesis and lipid metabolism14. Thus, the enriched biological terms found in the ovary-specific sub-network in this study indicate that the critical ovarian activities include steroidogenesis and lipid metabolism and that the actions of those DEGs in those biological terms play major roles in the regulation of the oestrous cycle and ovarian function for porcine prolificacy.
In addition, we determined the localization of genes involved in steroid biosynthesis found in the ovary-specific network and eicosanoid metabolism found in the endometrium-specific network to understand their cellular localization of expression and function in the reproductive tissues. The expression of CYP7A1 and CYP19A1 was mainly localized to ovarian GCs and the expression of CYP17A was localized to TCs, indicating that the steroidogenic activity in the ovary is mediated by the actions of TC and GC. The mRNA expression levels of CYP17A1 and CYP19A1 were great during the proestrus phase. In the endometrium, PTGS2 and PTGER2 expression was localized to endometrial epithelial and stromal cells with greatest abundance at late diestrus and proestrus phase, indicating that the endometrial production of PGs is active and PG action on the endometrium may be critical for endometrial function and oestrous cyclicity.
Immune signalling and regulation of porcine reproduction
There were also significantly enriched KEGG terms in the network that are related to immune signalling, biological oxidation, and the cells and molecules involved in local acute inflammatory response (Fig. 3). The dendritic cell regulation of TH1-TH2 development through antigen-specific T-cell signalling is an important mechanism in the immune response35. Eicosanoid metabolism was also significantly enriched in both the main network and the endometrium-specific sub-network (Fig. 4D). Prostaglandins are one of the multiple subfamilies of eicosanoids. They play a major role in inflammation and other oxidative stresses36,37. PGF2α secretion can act to lower P4 secretion, which is the signal that pregnancy has failed in pig. These findings demonstrate that the endometrium is directly involved in the biological reaction to a failed pregnancy during the oestrous cycle and involves prostaglandin and P4 signalling25. Moreover, a comparison of transcriptomes between pregnant and non-pregnant pigs reveals significant DEGs related to immune processes and pathways38, supporting our findings. In humans, after the withdrawal of progesterone induces menstruation, immune and haemostatic factors interact to induce endometrial haemostasis via platelet aggregation, fibrin deposition, and thrombus formation. Consequently, the cessation of menstrual bleeding can be achieved by the control of endometrial haemostasis39. Our results suggest that haemostasis and immune signalling are also important modulatory processes in pig and play a functional role in the endometrium during the oestrous cycle in non-pregnant pigs.
Developmental origin of the three tissues, oestrus cycling and the core network
The oviduct has a developmental origin common to endometrium and is sensitive to changes in ovarian-derived steroid hormones40. The oviductal transcriptome in embryo maturation and transport suggested that the oviduct is associated with progesterone signalling and showed that focal adhesions were enriched in the KEGG enrichment analysis18. Focal adhesions are integrin receptor-mediated macromolecular complexes that are associated with the extracellular matrix (ECM)41. In mammals, the focal adhesions connected with ECM receptors are key mechanisms in pregnancy recognition signalling after oocytes fertilised in the oviduct enter the uterus for gestation. The mechanisms of pregnancy recognition signalling sustain the functional lifespan of the CL, which produces progesterone21. Our results with non-pregnant pigs indicated that focal adhesions and ECM-receptor interactions were significantly enriched in the entire network (Fig. 3) as well as the oviduct-specific sub-network (Fig. 4F). The genes in the oviduct-specific sub-network were downregulated throughout the oestrous cycle and their expression pattern was exactly opposite to the pattern of progesterone secretion from the CL. These results suggest that the downregulation of genes in the oviduct is related to the negative response to pregnancy recognition signalling during the oestrous cycle in non-pregnant pigs. Thus, this result may be opposite in pregnant pigs. This may also affect the expression levels of DEGs of the core network connecting the tissue-specific networks. These DEGs may relate to the synchronization of the tissues. Alternatively, pregnancy may induce larger differences due to the different roles of the tissues in pregnancy.
The core network of bridging genes connecting the transcriptomes of three tissues regulating pig oestrus cycling
Based on the genes bridging among the three reproductive tissues (Fig. 5) and their significant GO terms (Table 1), the connectivity between the tissues and the shared biological roles of the three tissues were investigated. Ovarian steroids and cytokines regulate endometrial gene expression and oviduct function during the oestrous cycle and pregnancy10,42. CYP17A1, FDPS, HSD17B11, and TSPO were among the genes in the network that are involved in steroid metabolic processes. In particular, CYP17A1 is known to play a role in steroid hormone synthesis and a recent study that investigated the proteome of the ovine ovary suggested that upregulation of CYP17A1 is responsible for large litter size22. In pigs, CYP17A1 was also highly expressed in the endometrium during implantation23. On the other hand, CYP17A1 was downregulated, along with HSD17B11 and TSPO, during the oestrous cycle of non-pregnant pigs, as validated by qPCR (Fig. 6A). Moreover, other kinds of CYP (cytochrome P450) genes were also significantly downregulated in our study. The core network also revealed significant GO terms for cell adhesions and ECM. VCAN, which is a major component of the ECM, was commonly found in the GO terms. VCAN activates downstream cytokines such as IL6 and TNF through TLR2 and CD14 and these cytokine changes are important embryo-maternal signals in the porcine female reproductive tract during pregnancy43,44,45. In our study, genes such as the TLRs (Fig. 6A) and VCAN were significantly downregulated. Therefore, it can be implied that these genes have negative regulatory effects during the oestrous cycle in the reproductive tracts of non-pregnant pigs. We also focused on the high over-expression of FXDY4 in the endometrium throughout the oestrous cycle (Fig. 6A). A previous study compared transcriptomes during pregnancy and non-pregnancy on day 12 in the endometrium and indicated significantly up- and downregulated DEGs. Among them, FXDY4 was extremely downregulated during pregnancy [64]. In our study, we showed that FXDY4 was highly upregulated with the highest expression occurring on day 9. Therefore, the opposite regulation profiles of genes such as CYP17A1 and FXDY4 represented different changes in hormonal and immune signalling of reproductive tissues during pregnancy and non-pregnancy throughout the oestrous cycle. Therefore, we suggest further studies to clarify the molecular functions of these genes so they may be used as molecular markers.
Summarizing, the core network connecting the transcriptomes of the ovary, the oviduct, and the endometrium is enriched for two types of genes: (1) Steroid metabolism probably related to steroid hormone expression regulation, and (2) Communication between cells: ECM, cell adhesion, and immune processes.
Conclusions
What can we learn from our results about the regulation of tissue synchronization of three reproductive tissues in non-pregnant pigs throughout the oestrous cycle? First of all, the DEGs of the core network showed less differential expression than the DEGs differing among oestrus phases within each tissue. This suggests that the progression through the oestrus cycle in each tissue is regulated through regulation of gene expression. The results suggest that tissue synchronization may either require less regulation – e.g. because it is an intrinsic trait – or that it is regulated outside the core network. Remarkably, the genes of the core network mainly relate to steroid metabolism and regulation of (physical and immune signaling) cellular contacts. This indeed suggest that steroid hormones may be involved, and that direct interactions among cells inducing cellular signalling in each of the tissues are the main regulatory mechanism regulating the events of different tissues during the progression of the oestrus cycle.
Materials and Methods
Animals and tissue preparation
All experimental procedures involving animals were conducted in accordance with the Guide for Care and Use of Animals in Research and approved by the Institutional Animal Care and Use Committee of the National Institute of Animal Science (No. 2015-137). Twenty-one crossbred (Landrace × Yorkshire) gilts of similar age (6–8 months) and weight (100–120 kg) were utilised in this study after experiencing at least two oestrous cycles of normal duration (18–22 days). Gilts in the presence of boars were observed daily for oestrous behaviour. The day on which oestrous behaviour was first exhibited was designated as day 0. The ovary, endometrium, and oviduct reproductive tissues of gilts were collected on days 0 (the onset of oestrous behaviour), 3, 6, 9, 12, 15, or 18 of the oestrous cycle (n = 3 gilts/day) by hysterectomy to encompass the whole stage of the oestrous cycle (Fig. 1A). Endometrial tissue was dissected free of myometrium from the middle portion of the uterine horns. Oviductal tissue was collected from the ampulla region and a whole ovary on one side was collected as previously described46,47. Tissues were snap-frozen in liquid nitrogen and stored at −80 °C for RNA extraction. For in situ hybridisation analysis, cross sections of endometrium, oviduct, and ovary were fixed in 4% paraformaldehyde in PBS (pH 7.4) for 24 h and then embedded in paraffin, as previously described47.
Library preparations and sequencing
Total RNA was extracted from the endometrial, oviductal and ovarian tissues using TRIzol reagent (Invitrogen, Life Technology, Carlsbad, CA) according to the manufacturer’s recommendations. The quantity of RNA was assessed spectrophotometrically, and integrity of RNA was validated following electrophoresis in 1% agarose gel. The mRNA of 1 µg of total RNA was converted into a library of template molecules suitable for subsequent cluster generation using the reagents provided in the Illumina® TruSeq™ RNA Sample Preparation Kit. The first step in the workflow involved purifying the poly‐A containing mRNA molecules using poly‐T oligo‐attached magnetic beads. Following purification, the mRNA was fragmented into small pieces using divalent cations under elevated temperature. The cleaved RNA fragments are copied into first strand cDNA using reverse transcriptase and random primers. This is followed by second strand cDNA synthesis using DNA Polymerase I and RNase H. These cDNA fragments then go through an end repair process, the addition of a single ‘A’ base, and then ligation with adapters. The products were then purified and enriched with PCR to create the final cDNA library. The libraries were quantified using qPCR according to the qPCR Quantification Protocol Guide (Manufacturer) and qualified using an Agilent Technologies 2100 Bioanalyzer. The cDNA libraries were sequenced using the paired-end sequencing by Illumina HiSeq. 2000.
RNA-seq data processing and differentially expressed gene (DEG) analysis
In total, 130 million paired-end sequence reads were produced, with an average of 19.5 million reads per sample. The all of raw RNA-seq data were deposited at the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under the accession GSE108570. Before the RNA-seq data analysis, raw reads were processed for quality filtering by fastQC and the first 13 bp of each read were trimmed based on the Per Base Sequence Content from fastQC48. The standard Tuxedo protocol was followed to estimate the normalised expression levels of transcripts49. All trimmed reads were mapped against the reference genome (Sus scrofa 10.2, GCA_000003025.4) and transcriptome (Ensembl v78) from the Ensembl genome browser (http://www.ensembl.org/Sus_scrofa/) by tophat2 v2.0.13 using the default options. Mapping statuses were plotted and analysed by MultiQC v0.8dev50. All possible transcripts were inferred using the Reference Guided Transcriptome Assembly (RABT) mode in cufflinks v2.2.151. The gene expression level was obtained using the measurement of fragments per kilobase per million reads (FPKM). Geometric normalisation methods were used because at least two samples from different pigs were collected for each time point52. Differentially expressed genes (DEGs) were selected by Cuffdiff in the Tuxedo protocol with a q-value cut-off of 0.05 by comparing all tissues at the same time point. Principal component analysis (PCA) was performed to analyse relationships between individual samples and we employed ggfortify and cluster packages in R to visualise PCA results. Additional k-means clustering and PAM (Partitioning Around Medoids) algorithms were adopted because individuals were clearly clustered into three groups53.
For expression profiling across multiple tissues and periods of the oestrous cycle, further analyses were performed using in-house R scripts. The correlation values between tissues were calculated by Pearson Correlation using a log2 (FPKM + 1) value. Then, the complete-linkage clustering was performed and visualised with an FPKM frequency histogram. Genes that had fold changes (FC) of 2, 4, 8, 16, 32, 64, 128, and 256 were selected and the genes with an 8-FC showing high connectivity among tissues were visualised and analysed54. Z-score-transformed log2 (FPKM + 1) values were used to compare and analyse gene expression levels.
GCN analysis and visualisation
The GCN analysis was performed on transcripts with cut-off proceeded as follows: (1) the transcripts were removed when they did not have a significant FDR (q < 0.05) in any of the seven oestrous time points and three tissues; (2) the rRNA transcripts and de novo transcripts that had more than two gene symbols were removed; (3) a stringent significant level in DEG (absolute log2 FC ≥ 3.0) was employed to enhance the efficiency of network construction. As a consequence, only the transcripts that had absolute log2 FC greater than 3.0 in at least one of the seven oestrous time points and one of the three tissues were used for GCN analysis. The significant DEGs were entered in the PCIT algorithm31. The PCIT algorithm determined co-expression between genes using the concept of partial correlation coefficients with information theory. Thereafter, the PCIT algorithm has been used to establish significant connections (edges) and construct the network. Connections between two genes with a correlation estimate for co-expression between zero and one. We created a network by taking absolute co-expression correlations greater than 0.90 among those determined to be significant by the PCIT algorithm. Cytoscape version 3.4.0 was used to visualise the co-expression network55. In the topological view of the network, genes (nodes) were closer together when they had more common neighbours than the others.
Sub-networks were constructed based on the gene clustering analysis with visual identifications. The k-means clustering algorithm was used in the Multi Experiment Viewer software after median centring by narrowing down the optimal number of clusters with 1 K iterations56. The clustered genes had distinct expression patterns between tissues based on their log2 FC of FPKM across all phases of the oestrous cycle. Genes that had connections to at least two different tissues, designated bridging genes, were used to investigate the core-network responsible for connections between tissue-specific networks. Otherwise, genes were excluded when they were embedded within the tissue-specific networks or located outside of sub-networks. Bridging genes were used to rebuild the core network, which was visualised using the Prefuse force directed layout in the Cytoscape software. Then, four sub-clusters were identified that had a high number of connections in the core network. The same colours were always used in the figures to represent the different tissues (red, endometrium; yellow, ovary; and blue, oviduct) and oestrous cycle time points (red, D3; orange, D6; yellow, D9; green, D12; blue, D15; and purple, D18).
In silico functional analyses
The genes in the network were analysed for functional enrichment of Gene Ontology (GO) terms in biological processes and pathways using the DAVID Bioinformatics Resources 6.757. Major databases, including BBID, BIOCARTA, KEGG, PANTHER, and REACTOME, were used for the pathway enrichment analysis. The significantly enriched pathways were represented by fold enrichment and a –log10 P-value. GO annotations in biological processes were also enriched using the FAT option, which is a specific GO category filter in DAVID.
The enrichment analysis of pathways in DAVID was performed again for each of the clearly separated tissue-specific sub-networks of different tissues using the same method as described above. GO enrichment analyses for the sub-networks were also carried out by the BiNGO plugin in Cytoscape58. Then, the REVIGO visualisation tool was applied to make tree maps for the enriched GO terms59. The enrichment analyses using BiNGO were conducted with default conditions and with the basal significance level (P < 0.05). The data were analysed using the human annotation (using HUGO official gene symbols) and the pig annotation, separately. The core network, consisting of genes connecting the tissue-specific sub-networks (called “bridging genes”), functional annotation clustering in DAVID was performed to determine the various biological roles of core-network genes in all aspects of GO terms including biological processes, cellular components, and molecular functions.
Quantitative real-time PCR (qPCR)
The results of RNA-seq analysis were validated by qPCR with the same RNA samples as used for RNA-seq. qPCR was performed with the Applied Biosystems StepOnePlus System (Applied Biosystems, Foster City, CA, USA) using the SYBR Green method. Complementary DNAs (cDNAs) were synthesised from 4 µg of total RNA isolated from the tissues. Newly synthesised cDNAs (total volume of 21 μl) were diluted 1:4 with sterile water and then used for PCR. The Power SYBR Green PCR Master Mix (Applied Biosystems) was used for PCR reactions. The final reaction volume of 20 μl included 2 μl of cDNA, 10 μl of 2 × master mix, 2 μl of each primer, and 4 μl of dH2O. PCR conditions and sequences of primer pairs are listed in Supplementary data 1. The cycle thresholds determined for the target genes were normalised against the geometric mean of the reference genes, RPL7, UBB, and TBP, which have previously been used in porcine reproductive tissues47,60. Relative expression levels of the selected target genes were calculated using the 2−ΔΔCT method61. The identity of each amplified PCR product was verified by sequence analysis after cloning into the pCRII vector (Invitrogen). Regression analysis was performed to compare the expression values from the real-time PCR with RNA-seq results.
Non-radioactive in situ hybridisation
To determine the localisation of expression of selected genes in the reproductive tissues, a non-radioactive in situ hybridisation procedure was performed, as previously described with several modifications62. Sections (5 µm thick) were rehydrated through successive baths of xylene, 100% ethanol, 95% ethanol, diethylpyrocarbonate (DEPC)-treated water, and DEPC-treated PBS. Tissue sections were boiled in citrate buffer (pH 6.0) for 10 min. After washing in DEPC-treated PBS, sections were digested using 5 µg/ml Proteinase K (Sigma) in TE (100 mM Tris-HCl, 50 mM EDTA, pH 7.5) for 10 min at 37 °C. After post-fixation in 4% paraformaldehyde, tissue sections were incubated twice for 15 min each in PBS containing 0.1% active DEPC, and equilibrated for 15 min in 5 × saline sodium citrate (SSC). The sections were prehybridised for 2 h at 68 °C in hybridisation mix (50% formamide, 5 × SSC, 500 µg/ml herring sperm DNA, 250 µg/ml yeast tRNA). Sense and antisense riboprobes for each gene were generated using partial cDNAs cloned into pCRII vectors by linearising with appropriate restriction enzymes and labelling with digoxigenin (DIG)-UTP using a DIG RNA labelling kit (Roche, Indianapolis, IN, USA). The probes were denatured for 5 min at 80 °C and added to the hybridisation mix. The hybridisation reaction was carried out overnight at 68 °C. Sections hybridised with sense riboprobes served as negative controls. Pre-hybridisation and hybridisation reactions were performed in a box saturated with a 5 × SSC − 50% formamide solution to avoid evaporation. No coverslips were used. After hybridisation, sections were washed for 30 min in 2 × SSC at room temperature, 1 h in 2 × SSC at 65 °C, and 1 h in 0.1 × SSC at 65 °C. Probes bound to the section were detected immunologically using sheep anti-DIG Fab fragments covalently coupled to alkaline phosphatase and nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate (toluidine salt) as chromogenic substrate, in accordance with the manufacturer’s protocol (Roche).
References
Sharpe, R. M. & Franks, S. Environment, lifestyle and infertility–an inter-generational issue. Nature cell biology 4(Suppl), s33–40 (2002).
Soede, N. M., Langendijk, P. & Kemp, B. Reproductive cycles in pigs. Animal Reproduction Science 124, 251–258 (2011).
de Rijk, E. et al. Estrous cycle-dependent morphology in the reproductive organs of the female Gottingen minipig. Toxicologic pathology 42, 1197–1211 (2014).
Tortereau, A., Howroyd, P. & Lorentsen, H. Onset of puberty and normal histological appearances of the reproductive organs in peripubertal female Gottingen minipigs. Toxicologic pathology 41, 1116–1125 (2013).
Brown, J. B. Types of ovarian activity in women and their significance: the continuum (a reinterpretation of early findings). Human Reproduction Update 17, 141–158 (2011).
Spencer, T. E. & Bazer, F. W. Conceptus signals for establishment and maintenance of pregnancy. Reproductive biology and endocrinology: RB&E 2, 49–49 (2004).
Almiñana, C. et al. The battle of the sexes starts in the oviduct: modulation of oviductal transcriptome by X and Y-bearing spermatozoa. BMC genomics 15, 1–11 (2014).
Fazeli, A., Affara, N. A., Hubank, M. & Holt, W. V. Sperm-induced modification of the oviductal gene expression profile after natural insemination in mice. Biology of reproduction 71, 60–65 (2004).
Gandolfi, F., Brevini, T. A., Cillo, F. & Antonini, S. Cellular and molecular mechanisms regulating oocyte quality and the relevance for farm animal reproductive efficiency. Revue scientifique et technique (International Office of Epizootics) 24, 413–423 (2005).
Hunter, R. H., Cook, B. & Poyser, N. L. Regulation of oviduct function in pigs by local transfer of ovarian steroids and prostaglandins: a mechanism to influence sperm transport. Eur J Obstet Gynecol Reprod Biol 14, 225–232 (1983).
Hunter, R. H. F. Ovarian programming of gamete progression and maturation in the female genital tract. Zoological Journal of the Linnean Society 95, 117–124 (1989).
López-Úbeda, R et al. Oviductal Transcriptome Is Modified after Insemination during Spontaneous Ovulation in the Sow. PLoS One 10 (2015).
López-Úbeda, R. et al. The oviductal transcriptome is influenced by a local ovarian effect in the sow. Journal of Ovarian Research 9, 44 (2016).
Zhang, X. et al. Transcriptomic Analysis of Ovaries from Pigs with High And Low Litter Size. PLoS ONE 10, e0139514 (2015).
Samborski, A. et al. Transcriptome changes in the porcine endometrium during the preattachment phase. Biology of reproduction 89, 134 (2013).
Kim, M. et al. Analysis of Stage-Specific Gene Expression Profiles in the Uterine Endometrium during Pregnancy in Pigs. PLoS ONE 10, e0143436 (2015).
Ruiz-Alonso, M., Blesa, D. & Simón, C. The genomics of the human endometrium. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1822, 1931–1942 (2012).
Hess, A. P. et al. The human oviduct transcriptome reveals an anti-inflammatory, anti-angiogenic, secretory and matrix-stable environment during embryo transit. Reproductive biomedicine online 27, 423–435 (2013).
Kadir, R. A. & Davies, J. Hemostatic disorders in women. Journal of Thrombosis and Haemostasis 11, 170–179 (2013).
Minge, C. E., Robker, R. L. & Norman, R. J. PPAR Gamma: Coordinating Metabolic and Immune Contributions to Female Fertility. PPAR Research 2008, 243791 (2008).
Bazer, F. W. et al. Uterine biology in pigs and sheep. Journal of Animal Science and Biotechnology 3, 23 (2012).
Miao, X., Luo, Q., Zhao, H. & Qin, X. Ovarian proteomic study reveals the possible molecular mechanism for hyperprolificacy of Small Tail Han sheep. Scientific Reports 6, 27606 (2016).
Chen, X. et al. Differential Gene Expression in Uterine Endometrium During Implantation in Pigs. Biology of reproduction 92(52), 51–14 (2015).
Kurita, T. et al. Paracrine regulation of apoptosis by steroid hormones in the male and female reproductive system. Cell Death And Differentiation 8, 192 (2001).
Blatchley, F. R., Walker, F. M. & Poyser, N. L. P. prostaglandin F2alpha, and oestradiol in the utero-ovarian venous plasma of non-pregnant and early, unilaterally pregnant guinea-pigs. J Endocrinol 67, 225–229 (1975).
Henricks, D. M., Guthrie, H. D. & Handlin, D. L. Plasma Estrogen, Progesterone and Luteinizing Hormone Levels During the Estrous Cycle in Pigs. Biology of reproduction 6, 210–218 (1972).
Downey, B. R. Regulation of the Estrous Cycle in Domestic Animals — A Review. The Canadian Veterinary Journal 21, 301–306 (1980).
Yang, Y. et al. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nature communications 5, 3231 (2014).
Zhao, W. et al. Weighted gene coexpression network analysis: state of the art. Journal of biopharmaceutical statistics 20, 281–300 (2010).
Pérez-Montarelo, D. et al. Porcine Tissue-Specific Regulatory Networks Derived from Meta-Analysis of the Transcriptome. PLOS ONE 7, e46159 (2012).
Reverter, A. & Chan, E. K. F. Combining partial correlation and an information theory approach to the reversed engineering of gene co-expression networks. Bioinformatics 24, 2491–2497 (2008).
Sanderson, J. T. The Steroid Hormone Biosynthesis Pathway as a Target for Endocrine-Disrupting Chemicals. Toxicological Sciences 94, 3–21 (2006).
Hansen, M., Flatt, T. & Aguilaniu, H. Reproduction, Fat Metabolism, and Lifespan – What Is the Connection? Cell metabolism 17, 10–19 (2013).
Heine, P. A., Taylor, J. A., Iwamoto, G. A., Lubahn, D. B. & Cooke, P. S. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proceedings of the National Academy of Sciences of the United States of America 97, 12729–12734 (2000).
Moser, M. & Murphy, K. M. Dendritic cell regulation of TH1-TH2 development. Nat Immunol 1, 199–205 (2000).
Ricciotti, E. & FitzGerald, G. A. Prostaglandins and Inflammation. Arteriosclerosis, thrombosis, and vascular biology 31, 986–1000 (2011).
Basu, S. Bioactive eicosanoids: Role of prostaglandin F2α and F2-isoprostanes in inflammation and oxidative stress related pathology. Molecules and Cells 30, 383–391 (2010).
Franczak, A., Wojciechowicz, B. & Kotwica, G. Transcriptomic analysis of the porcine endometrium during early pregnancy and the estrous cycle. Reproductive biology 13, 229–237 (2013).
Davies, J. & Kadir, R. A. Endometrial haemostasis and menstruation. Reviews in Endocrine and Metabolic Disorders 13, 289–299 (2012).
Menezo, Y. & Guerin, P. The mammalian oviduct: biochemistry and physiology. Eur J Obstet Gynecol Reprod Biol 73, 99–104 (1997).
Zaidel-Bar, R., Itzkovitz, S., Ma’ayan, A., Iyengar, R. & Geiger, B. Functional atlas of the integrin adhesome. Nature cell biology 9, 858–867 (2007).
Bazer, F. W. et al. Novel pathways for implantation and establishment and maintenance of pregnancy in mammals. Molecular Human Reproduction 16, 135–152 (2010).
Kim, S. et al. Carcinoma Produced Factors Activate Myeloid Cells via TLR2 to Stimulate Metastasis. Nature 457, 102–106 (2009).
Modrić, T., Kowalski, A. A., Green, M. L., Simmen, R. C. M. & Simmen, F. A. Pregnancy-dependent Expression of Leukaemia Inhibitory Factor (LIF), LIF Receptor-β and Interleukin-6 (IL-6) Messenger Ribonucleic Acids in the Porcine Female Reproductive Tract. Placenta 21, 345–353 (2000).
Ashworth, M. D., Ross, J. W., Stein, D., White, F. & Geisert, R. D. Endometrial gene expression of acute phase extracellular matrix components following estrogen disruption of pregnancy in pigs. Anim Reprod Sci 122, 215–221 (2010).
Buhi, W. C., Vallet, J. L. & Bazer, F. W. De novo synthesis and release of polypeptides from cyclic and early pregnant porcine oviductal tissue in explant culture. The Journal of experimental zoology 252, 79–88 (1989).
Seo, H., Choi, Y., Shim, J., Yoo, I. & Ka, H. Comprehensive analysis of prostaglandin metabolic enzyme expression during pregnancy and the characterization of AKR1B1 as a prostaglandin F synthase at the maternal-conceptus interface in pigs. Biology of reproduction 90, 99 (2014).
Andrews, S. FastQC. A quality control tool for high throughput sequence data. Babraham Bioinformatics. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2014).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protocols 7, 562–578 (2012).
Ewels, P., Magnusson, M., Lundin, S., Kaller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics (2016).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotech 28 (2010).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol 11 (2010).
Kaufman, L., Rousseeuw, P. J. Partitioning Around Medoids (Program PAM). In: Finding Groups in Data. John Wiley & Sons, Inc (2008).
Lin, S., et al. Comparison of the transcriptional landscapes between human and mouse tissues. Proc Natl Acad Sci 111 (2014).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003).
Howe, E. A., Sinha, R., Schlauch, D. & Quackenbush, J. RNA-Seq analysis in MeV. Bioinformatics 27, 3209–3210 (2011).
Huang, D. W. et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biology 8, R183–R183 (2007).
Saito, R. et al. A travel guide to Cytoscape plugins. Nat Meth 9, 1069–1076 (2012).
Supek, F., Bošnjak, M., Škunca, N. & Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6 (2011).
Martínez-Giner, M., Noguera, J. L., Balcells, I., Fernández-Rodríguez, A. & Pena, R. N. Selection of Internal Control Genes for Real-Time Quantitative PCR in Ovary and Uterus of Sows across Pregnancy. PLoS ONE 8, e66023 (2013).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Braissant, O. & Wahli, W. Differential expression of peroxisome proliferator-activated receptor-alpha, -beta, and -gamma during rat embryonic development. Endocrinology 139, 2748–2754 (1998).
Acknowledgements
This study was carried out with the support of 2017 the RDA Fellowship Program of the NIAS and “Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ01119103 and PJ00806802), Rural Development Administration, Republic of Korea. The English in this document has been checked by at least two professional editors, both native speakers of English. For a certificate, please see: http://www.textcheck.com/certificate/ieW95k.
Author information
Authors and Affiliations
Contributions
J.M.K. analysed the data and wrote the paper. J.E.P., I.Y., J.H., N.K., E.S.C. and B.C. performed the experiment, provided text and data for the methods, and contributed comments on the manuscript. M.t.P., W.J.L. and T.H.K. participated in analysis of the data and writing of the manuscript. S.C. and H.K. designed and supervised the experiment. K.T.L. supervised the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, JM., Park, JE., Yoo, I. et al. Integrated transcriptomes throughout swine oestrous cycle reveal dynamic changes in reproductive tissues interacting networks. Sci Rep 8, 5436 (2018). https://doi.org/10.1038/s41598-018-23655-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23655-1
This article is cited by
-
Hormone-regulated PKA activity in porcine oviductal epithelial cells
Cell and Tissue Research (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
