
Abstract
Dimorphic fungal pathogens cause a significant human disease burden and unlike most fungal pathogens affect immunocompetent hosts. To examine the origin of virulence of these fungal pathogens, we compared genomes of classic systemic, opportunistic, and non-pathogenic species, including Emmonsia and two basal branching, non-pathogenic species in the Ajellomycetaceae, Helicocarpus griseus and Polytolypa hystricis. We found that gene families related to plant degradation, secondary metabolites synthesis, and amino acid and lipid metabolism are retained in H. griseus and P. hystricis. While genes involved in the virulence of dimorphic pathogenic fungi are conserved in saprophytes, changes in the copy number of proteases, kinases and transcription factors in systemic dimorphic relative to non-dimorphic species may have aided the evolution of specialized gene regulatory programs to rapidly adapt to higher temperatures and new nutritional environments. Notably, both of the basal branching, non-pathogenic species appear homothallic, with both mating type locus idiomorphs fused at a single locus, whereas all related pathogenic species are heterothallic. These differences revealed that independent changes in nutrient acquisition capacity have occurred in the Onygenaceae and Ajellomycetaceae, and underlie how the dimorphic pathogens have adapted to the human host and decreased their capacity for growth in environmental niches.
Similar content being viewed by others


Introduction
Among millions of ubiquitous fungal species that pose no threat to humans, a small number of species cause devastating diseases in immunocompetent individuals. Notably, many human pathogenic species are ascomycetes in the order Onygenales, which comprises dermatophytes (e.g. Trichophyton spp.), the spherule-forming dimorphic pathogen Coccidioides, and yeast-forming dimorphic pathogens Histoplasma, Paracoccidioides, and Blastomyces. Dimorphism is a specialized morphogenetic adaptation allowing both growth in the environment and colonization of a host and is critical for the lifecycle of dimorphic fungal pathogens1. In yeast-forming pathogens, dimorphism has been broadly defined as the ability to generate both yeast (e.g. blastoconidia) in the host or at 37 °C and mycelia (e.g. conidia-producing or other hyphae) in the environment1. During fungal evolution, dimorphism has arisen independently multiple times in saprophytic fungi, as is manifested in the wide distribution of dimorphic species across the Ascomycota, Basidiomycota and Zygomycota phyla2. Notably, the dimorphic fungi in the order Onygenales are primary pathogens causing systemic mycosis in healthy humans and collectively cause over 650,000 new infections a year in the United States alone3. Dimorphic fungi can persist as latent infections in tens of millions of people worldwide and may reactivate when the host becomes immune-deficient; symptoms of active infection include pneumonia, acute respiratory distress syndrome, and disseminated disease, which can affect multiple organ systems1.
The largest cluster of thermally dimorphic human pathogenic fungi belongs to the onygenalean family Ajellomycetaceae, which includes Histoplasma, Paracoccidioides, and Blastomyces. In addition to these medically important genera, the Ajellomycetaceae family also includes more rarely observed pathogenic species including Emmonsia parva and Ea. crescens, which undergo a thermal dimorphic transition to produce adiaspores rather than yeast and cause adiospiromycosis sporadically in humans4,5. This group also includes Lacazia loboi, the yeast-like etiological agent of lobomycosis6. Of public health concern, recent reports have documented the worldwide emergence of yeast-forming Ajellomycetaceae that cause systemic mycoses predominantly in immunocompromised patients, often with high case-fatality rates7,8. Morphological and phylogenetic analyses were combined to demarcate species boundaries within the Ajellomycetaceae with clinical significance, which led to a proposed revision of the taxonomy; this included addition of the new genus Emergomyces, which includes the new species Es. africanus and Es. orientalis, the description of the new species Blastomyces percursus, and ongoing efforts to define additional Emmonsia-like and Blastomyces-like species8,9,10. Dimorphic species within the Ajellomycetaceae are typically restricted to specific ecological niches and geographical regions. For example, while H. capsulatum and Es. pasteurianus are considered to have worldwide distribution, B. dermatitidis, P. brasiliensis, and Es. africanus are restricted to North America, Latin America and South Africa, respectively. Non-dimorphic species have also been documented as early diverging Ajellomycetaceae, including Emmonsiellopsis, Helicocarpus griseus and Polytolypa hystricis, which have not been associated with disease in mammals. These species are found in environmental samples and do not appear to undergo a thermally regulated dimorphic transition11,12,13.
We hypothesized that such phenotypic differences within the Ajellomycetaceae could be associated with gene family expansions or contractions, as a consequence of gene duplication and gene loss events, and that species comparison might reveal evolutionary mechanisms of adaptation in the systemic dimorphic fungi. Previous comparative and population genomic studies within Ajellomycetaceae had found evidence of gene family contractions and expansions associated with virulence5,14,15. Other Onygenales genera outside the Ajellomycetaceae represented by published genome sequences include Coccidioides and the non-pathogenic related species Uncinocarpus reesii16,17, as well as diverse dermatophytes that cause skin infections18,19. Recently, the genomes of four non-pathogenic Onygenaceae species closely related to Coccidioides were described, providing additional resolution into changes in gene content between pathogenic and non-pathogenic Onygenales20. Together these studies helped establish how gene content is related to the life cycle of different dimorphic pathogenic fungi and dermatophytes; for example, gene family contractions in cellulases and other plant metabolism genes, and gene family expansions in proteases, keratinases and other animal tissue metabolism genes, indicated that dimorphic fungi switched from a nutrition system based on plants to a system based on animals, though mostly relative to outgroups to the Onygenales such as Aspergillus14,17,18,19. In the Onygenales, this hypothesis was tested experimentally; comparing growth on a wide variety of compounds revealed that in Uncinocarpus reesii hyphal growth was restricted on carbohydrates and considerably improved on proteins14.
To examine key transition points in evolution of virulence and host adaptation in the dimorphic fungi, we increased the phylogenetic density within the Ajellomycetaceae by sequencing the genomes of non-pathogenic and emerging species and then performed comparative genomic analyses of the systemic, opportunistic, and non-pathogenic species. Although representative genomes for most of the described pathogenic Onygenales genera have been sequenced, the only strictly non-pathogenic species that have been sequenced are closely related to Coccidioides in the family Onygenaceae. Here, we have sequenced four additional Onygenales species from the family Ajellomycetaceae; this includes H. griseus and P. hystricis, neither of which has been associated with disease in mammals, and two additional adiaspore-forming strains of Ea. parva and Ea. crescens with distinct phenotypic properties. Leveraging our previous genomic studies and these additional genome sequences, we have characterized the extent of gene family expansions and contractions to provide a better understanding of the evolutionary mechanisms of adaptation in systemic dimorphic fungi in the Ajellomycetaceae.
Results
Genome comparisons of Ajellomycetaceae, the largest group of dimorphic human pathogenic fungi
Building on previous genomic analyses of the members from the Ajellomycetaceae5,9,14,15,21, we sequenced using Illumina technology the genomes of four species within Ajellomycetaceae with demarcated phenotypic differences. In addition to Emmonsia parva UAMH139 and Emmonsia crescens UAMH3008, we sequenced the genomes of one strain of Emmonsia parva UAMH130 (type strain) and one additional strain of Ea. crescens UAMH4076. Ea. parva UAMH130 is from lungs of a rodent in USA, and Ea. crescens UAMH4076 is from a greenhouse source in Canada22. We also sequenced two species that live in soil, where they are found in animal excrement but not associated with disease; neither species appears to undergo a thermal dimorphic switch to a pathogenic phase, as there is no observation of a yeast-like form13,22. This includes one strain of Helicocarpus griseus, UAMH5409 (type strain; synonym Spiromastix grisea), isolated from gazelle dung in Algeria, and one strain of Polytolypa hystricis, UAMH7299, isolated from porcupine dung in Canada.
The de novo genome assemblies of these four species have similar sizes to those of other genera within the Ajellomycetaceae including Histoplasma, Emergomyces and Paracoccidioides5,9,14,15 and have high representation of conserved eukaryotic genes (Figures S1, S2). The assembly sizes were 27.8 Mb in Ea. parva UAMH130, 33.8 Mb in Ea. crescens UAMH4076, 32.0 Mb in P. hystricis, and 34.7 Mb in H. griseus (Table 1). In addition, using both the CEGMA23 and BUSCO24 conserved eukaryotic gene sets, we found that the predicted genes for each sequenced species are nearly complete; all of these genomes include 96–98% of these core gene sets (Figure S2). The total numbers of predicted genes in Ea. parva UAMH130 (n = 8,202), and Ea. crescens UAMH4076 (n = 8,909) were similar to those found in annotated genome assemblies of other Ajellomycetaceae (n = 9,041 on average); however, relative to other Ajellomycetaceae, P. hystricis and H. griseus have a modest increase in gene content (n = 9,935 and n = 10,225, respectively; Table 1; Figure S1).
Examining the guanine-cytosine (GC) content in the genomes Ea. parva UAMH130, Ea. crescens UAMH4076, H. griseus UAMH5409 and P. hystricis UAMH7299 revealed that none of these genome assemblies showed evidence of the pronounced bimodality of GC-content observed in B. gilchristii and B. dermatitidis (Figure S3; Table 1;5). This highlights that the acquisition of repetitive elements contributing to bimodal GC distribution were a unique phenomenon during the evolution of the B. gilchristii and B. dermatitidis within the Onygenales. In addition, UAMH139 showed a small peak of low GC that is largely absent in UAHM130 (44.4% and 47.5% GC, respectively; Table 1), which is consistent with the phylogenetic divergence between these two strains, and highlights an independent gain of repetitive sequences (10.4% and 2.6% repetitive bases in UAMH139 and UAMH130, respectively; Table 1). Similarly, Ea. crescens UAMH3008 and UAHM4076 have demarcated differences in GC and repetitive content; UAHM4076 has a higher peak of low GC sequences (42.9% GC, 15.0% repetitive bases in UAMH4076; 45.2% GC, 5.1% repetitive bases in UAMH3008; Table 1). Additionally, the more basally branching strain P. hystricis UAMH7299 has a moderate representation of low GC sequences (45.6% GC, 12.8% repeat), but in H. griseus UAMH5409 these repetitive elements are almost absent (48.0% GC, 1.8% repeats; Figure S3). These results highlight the independent expansion of repetitive elements during Ajellomycetaceae evolution.
Phylogenomic analysis revealed multiple transitions within dimorphic pathogens to enable human infection
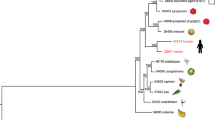
We estimated a strongly supported phylogeny of the newly sequenced Ea. parva UAMH130, Ea. crescens UAMH4075, H. griseus and P. hystricis strains relative to other dimorphic fungi using maximum likelihood and 2,505 single copy core genes (Fig. 1). Branch length and topology supported the placement and relationships of P. hystricis and H. griseus within the Ajellomycetaceae as basally branching. The two Ea. parva isolates (UAMH130 and UAMH139) do not form a single well-defined clade, confirming that Ea. parva may not be a single species. In addition to longer branch lengths suggesting higher divergence of these isolates, the average genome-wide identity between UAMH130 and UAMH139 is much lower even comparing across groups that represent recently subdivided species, for example, within B. gilchristii and B. dermatitidis or P. lutzii and P. brasiliensis, with an average identity of 88.6% between Ea. parva isolates versus 95.8% between Blastomyces species and 91.1% between Paracoccidiodies species (Figure S4). Ea. parva (type strain UAMH130) is the most basally branching member of the clade including Ea. parva UAMH139 and Blastomyces species B. percursus, B. gilchristii, and B. dermatitidis. Previous molecular approaches12,25 and recent multi-locus approaches8,9 also support the polyphyletic nature of Ea. parva isolates. Although both isolates of Ea. crescens (UAMH3008 and UAMH4076) form a single clade, the genetic variation between them is also greater than that observed within other species from the same genus in the Ajellomycetaceae, with long branch lengths and slightly lower average genome identity (91.8%) between these isolates than in other intraspecies comparisons within the family (Figure S4). The Ea. crescens clade is closely related to the recently proposed genus Emergomyces including the Es. pasteurianus/africanus species9. The Ea. crescens – Emergomyces clade is a sister group of the clade including Histoplasma and Blastomyces, and Paracoccidioides is in a basal position for the dimorphic pathogens of the Ajellomycetaceae (Fig. 1).

Phylogenomic relationships and diversity in the family Ajellomycetaceae. Maximum likelihood phylogeny (2,505 core genes based on 1,000 replicates) including 20 annotated genome assemblies of species within the family Ajellomycetaceae, five other Onygenales and three Aspergilli. All nodes were supported by 100% of bootstrap replicates. Color bars indicate the genus, and icons represent most common source or host. Illustrations of the most common morphology at 37 °C or in the host are depicted for selected species highlighting the phenotypic variation within the Ajellomycetaceae and the transition to human hosts represented by pathogenicity traits.
The intercalated clades of primary pathogens (e.g. H. capsulatum, P. brasiliensis and B. dermatitidis), opportunistic pathogens (e.g. Es. pasteurianus, Es. africanus) producing a parasitic yeast phase, and non-pathogens producing adiaspore-type cells rather than yeast cells (e.g. Ea. parva and Ea. crescens) suggests that the dimorphic fungi in the Ajellomycetaceae have undergone numerous evolutionary transitions allowing adaptation, infection and virulence to humans and other mammals, and that interactions with other eukaryotes in the environment may help maintain the capacity for pathogenic growth in a mammalian host (Fig. 1; Table 2). The non-thermally-dimorphic species H. griseus and P. hystricis are highly supported as early diverging lineages of Ajellomycetaceae as previously suggested13; genomic analysis strongly supports P. hystricis as the earliest diverging lineage within the sequenced Ajellomycetaceae and large genetic variation between these early saprophytes. The basal position of both H. griseus and P. hystricis in the family Ajellomycetaceae and the large divergence between them suggests that the history of this family could involve evolution from saprophytic non-thermotolerant and non-yeast-forming ancestral species to dimorphic human pathogenic species after the divergence with H. griseus, including the adaptation to higher temperatures and the appearance of a dimorphic switch and a yeast parasitic phase (Fig. 1).
Substantial gene family contractions underlie the shift from growth on plant to animals
By comparing the gene conservation and functional annotation of primary and opportunistic dimorphic pathogens with their saprophytic relatives H. griseus and P. hystricis, we identified significant decreases in gene family size (PFAM domain and GO-term counts, corrected p-value < 0.05) associated with a host/substrate shift from plants to animals in the pathogenic Ajellomycetaceae (Fig. 2). Plant cell wall degrading enzymes are reduced in number or totally absent in dimorphic pathogens compared with H. griseus and P. hystricis, including several glycosyl hydrolases, and fungal cellulose binding domain-containing proteins (Fig. 2; Table S2). These gene family changes suggest that dimorphic fungi from Ajellomycetaceae are not typical soil fungi in that they might maintain a close association with living mammals or other organisms such as protozoa or insects. This transition appears to have occurred within the family Ajellomycetaceae after divergence from Helicocarpus and Polytolypa (Fig. 1). Our analysis also recapitulates previous reports comparing smaller numbers of genomes from the order Onygenales5,14,17,20 that showed that genes coding for enzymes involved in the deconstruction of plant cell walls were absent from all the human pathogens (e.g. Paracoccidioides, Blastomyces, Histoplasma, Coccidioides) and non-pathogenic Onygenaceae (e.g. Uncinocarpus reesii, Byssoonygena ceratinophila, Amauroascus mutatus, Amauroascus niger, Chrysosporium queenslandicum) whereas these enzymes were commonly conserved in species outside the order Onygenales (including Aspergillus, Fusarium, Neurospora; Fig. 2; Table S2). Notably, increasing the phylogenetic density within the order Onygenales in this study revealed that the loss of plant cell wall degradative enzymes occurred independently in two families within the Onygenales, and this highlights transitions between saprophytic and pathogenic species, where Helicocarpus and Polytolypa have retained enzymes required to live on plant material in the soil, whereas non-pathogenic species from the Onygenaceae, which are not adapted to survive in mammals, have also lost capacity to degrade plant material.

Gene family changes across the Ajellomycetaceae. Heatmap depicting results of a protein family enrichment analysis (PFAM domains; corrected p-value < 0.05) comparing the gene content of the non-dimorphic/non-pathogenic Polytolypa hystricis and Helicocarpus griseus with all other depicted members of the family Ajellomycetaceae including the dimorphic pathogens Histoplasma, Emmonsia, Blastomyces, Emergomyces and Paracoccidioides. Values are colored along a blue (low counts) to red (high counts) color scale, with color scaling relative to the low and high values of each row. Each protein family domain has a color code (right) indicating a global functional category. Es: Emergomyces; Ea: Emmonsia.
Other notable changes in gene content included loss of protein families associated with secondary metabolite biosynthesis such as polyketide synthase dehydratases, and beta-ketoacyl synthases domains in dimorphic Ajellomycetaceae relative to H. griseus and P. hystricis (Fig. 2; Table S3). We identified predicted biosynthetic clusters using the program antiSMASH (antibiotics and Secondary Metabolism Analysis Shell26). Overall, dimorphic pathogenic fungi from Ajellomycetaceae have fewer polyketide synthase (PKS) gene clusters than the saprophytic H. griseus and P. hystricis (Table S3; Figure S5). Dimorphic pathogens have lost essential genes or complete clusters of the type 1 and type 3 PKS and terpene clusters (Table S3). Type 3 PKS are not common in fungi and were not previously reported in the Ajellomycetaceae. Furthermore, H. griseus and P. hystricis but not dimorphic fungi have genes related to those involved in antibiotic biosynthetic pathways (ko01055; Table S2). Previous studies have shown that P. hystricis secretes a pentacyclic triterpenoid exhibiting antifungal and antibiotic activity, denominated Polytolypin27, suggesting this terpene cluster is intact and may produce this molecule. Loss of such secondary metabolic pathways may weaken responses to other microbes in the environment. In addition to secondary metabolite biosynthesis, Ajellomycetaceae dimorphic fungi showed significant decreases in pathways related with the production of sphingolipids, chloroalkane and chloroalkene degradation, linoleic acid metabolism, and bisphenol degradation (KEGG-EC counts, corrected p-value < 0.05; Table S2). Loss of degradative pathways for bisphenol and chloralkanes in particular likely reflect encountering these molecules in the environment but not during pathogenic growth. Altogether, these shifts in gene content highlight how the Ajellomycetaceae dimorphic pathogenic fungi became less adapted to survive in the soil than the more basally diverging saprophytic species H. griseus and P. hystricis.
Transition among carbohydrate metabolism and protein catabolism
We found that the evolution of carbohydrate metabolism and protein catabolism also occurred independently within the Ajellomycetaceae. We annotated carbohydrate active enzymes (CAZy) and peptidases (MEROPS; Methods), and using enrichment analysis found substantial shifts in the relative proportion of these groups among dimorphic and saprophytic species. Notably, the pathogenic Ajellomycetaceae species, but not the saprophytic species H. griseus and P. hystricis, showed a dramatic reduction of carbohydrate active enzymes, including 44 categories that were totally absent and 29 that were significantly depleted; these included glycoside hydrolases, glycosyltransferases, carbohydrate esterases, and polysaccharide lyases (corrected p < 0.05; Fig. 3). The fact that these enzymes are reduced in dimorphic pathogenic Ajellomycetaceae but not in H. griseus and P. hystricis confirms that reduced carbohydrate metabolism occurred independently Ajellomycetaceae from the reduction in the Onygenaceae, originally noted in the spherule-dimorphic pathogen Coccidioides17. In contrast to carbohydrate active enzymes, we found that the total number of peptidases is relatively similar in dimorphic and saprophytic species within the Ajellomycetaceae. Significant protein family expansions were found to be specific in Blastomyces, Histoplasma and Onygenaceae and Arthrodermataceae species relative to P. hystricis and H. griseus, including the A11, S12, and S08A peptidases (corrected p < 0.05; Fig. 3). The peptidase family A11 contains endopeptidases involved in the processing of polyproteins encoded by retrotransposons, and the increase in copy number is correlated with the genome expansion due to the proliferation of repetitive elements in Blastomyces5. The peptidase family S12 contains serine-type D-Ala-D-Ala carboxypeptidases, and S8 contains the serine endopeptidase subtilisin. Two families of fungal metalloendopeptidases (S8 and M35) were previously reported to be expanded in Coccidioides relative to saprophytic species outside the order Onygenales17. In addition, it has been reported that the Ajellomycetaceae includes fewer copies of multiple classes of peptidases (M36, M43, S8) as well as an associated inhibitor (I9, inhibitor of S8 protease) relative to other Onygenales5. Comparing across all the Onygenales, we found that all pathogenic Onygenales, as well as saprophytes related to Coccidioides such as U. reesii, had a higher ratio of proteases to carbohydrate active enzymes than the saprophytic Ajellomycetaceae H. griseus and P. hystricis (Fig. 3). This strengthens the evidence that the adaptation and specialization of dimorphic Ajellomycetaceae occurred independently and that non-pathogenic Onygenaceae may represent an intermediate state in showing gene content changes associated with pathogens, which allowed these fungi to use proteins as a main source of energy in nutrient-limited environments.

Shifts in carbohydrate metabolism and peptidase activity in Ajellomycetaceae. Heatmaps depicting for each taxon carbohydrate metabolism categories (CAZy) and peptidase families (MEROPs) across the Ajellomycetaceae species included in this study and other compared genomes. These categories were found significantly enriched (corrected p-value < 0.05) in the non-dimorphic systemic fungi P. hystricis and H. griseus relative to Ajellomycetaceae dimorphic fungi, or within Ajellomycetaceae species and other compared genomes from Onygenaceae and Arthrodermataceae families or Eurotiales order. The ratio of MEROPS genes to CAZY genes for each genome across the Ajellomycetaceae species included in this study and other compared genomes is shown at right. Es: Emergomyces; Ea: Emmonsia.
Loss of transporters in Ajellomycetaceae dimorphic fungi
In addition to major shifts in enzymes that support nutrient acquisition, the dimorphic Ajellomycetaceae have undergone losses of families of transporters. These include the large classes of major facilitator superfamily transporters (PF07690) and sugar transporters (PF00083) (Fig. 2). The number of transporters correlates with the numbers of genes predicted to have transmembrane helices (1,848 and 1,745 proteins have predicted transmembrane domains in H. griseus and P. hystricis, respectively, relative to an average of 1,490 in dimorphic Ajellomycetaceae pathogens, Fig. 4). To characterize gene gains and losses in transporters, we annotated transporter families (Methods;28), and found significant expansion of 18 classes of transporters encompassing 9 families in H. griseus and P. hystricis relative to dimorphic Ajellomycetaceae pathogens (corrected p-value < 0.05; Fig. 4). Transporter families that were totally absent or significantly reduced in pathogenic species included the sugar porter, anion:cation symporter, AAA-ATPase, ammonium transporter and carnitine O-Acyl transferase. The substrates of these transporters include galacturonate, Rip1, hexoses, dipeptides, allantoate, ureidosuccinate, allantoin, glycerol, sulfate, sulfite, thiosulfate, sulfonates, tartrate, thiamine, cellobiose, cellodextrin, ammonia, methylamine, phenylacetate, beta-lactams, trichothecene, and carnitine O-octanoyl (Fig. 4). Most of these compounds are only present in the plant cell wall, including the cellulose, hemicellulose, or pectin types, supporting that dimorphic pathogens have evolved to have more limited compounds from plant cell wall as source of energy.

Shifts in metabolism matched substrate preference within Ajellomycetaceae contraction of transporters within the Ajellomycetaceae. (left) Total number of transporters identified in the genomes for each taxon included in this study. (right) Heatmap depicting the number of family transporters (TCDB; color-code blue: low, red: high) for each taxon across the Ajellomycetaceae species and other compared genomes. These families were found significantly enriched (test, corrected p-value < 0.05) in the non-dimorphic P. hystricis and H. griseus relative to dimorphic Ajellomycetaceae fungi. Es: Emergomyces; Ea: Emmonsia.
Pathogenesis-related genes are conserved in both saprophytic and pathogenic lifestyles
Genes known to be important in the response to stress imposed by the host, including virulence-associated or yeast-phase specific genes of central importance in dimorphic fungi in the Onygenales, are conserved in H. griseus and P. hystricis. These include heat shock response proteins (HSF, HSP90, HSP70), dimorphic switch related proteins (RYP1, RYP2, RYP3, AGS1, FKS1), oxidative stress and hypoxia response proteins (CATB, CATP, SOD3, SRB1), as well as antigens (PbGP43, PbP27, BAD1; Table S4;21). In dimorphic pathogenic fungi both morphological transitions and growth temperature are linked, and loss of genes necessary for high-temperature growth and morphological transition in these pathogens results in attenuated virulence, which highlights that they are essential for pathogenesis29,30,31. Some of these genes such as the RYP1-3 transcriptional regulators and the dimorphism-regulating histidine kinase DRK1 have been connected with morphogenetic adaptation in response to environmental stimuli and with key determinants of the mycelia to yeast transition in dimorphic human pathogenic fungi in the Ajellomycetaceae29,30. Although H. griseus and P. hystricis are not dimorphic and cannot grow at 37 °C, the heat shock response has been shown to be highly evolutionarily conserved among eukaryotes. The fact that genes of central importance for high-temperature growth and dimorphic switch are conserved in H. griseus and P. hystricis suggests that these saprophytes may sense different gradations of temperature, or that the mechanism that triggers a rapid response to temperature shifts may not be rapid enough to enable these fungi to grow at 37 °C.
Dimorphic Ajellomycetaceae may have evolved specialized mechanisms to regulate their transcriptional responses; these include expansion of proteins related to the regulation of transcription, and protein kinase activity (GO term enrichment analysis; q-value < 0.05; Table S2), which are all expanded in dimorphic pathogens relative to H. griseus and P. hystricis. Thus, the gain of transcription factors, transcriptional regulators and phosphotransferases suggests that rapid evolution of transcriptional mechanisms may underlie the adaptation to both high temperature and the dimorphic transition. As one of the largest categories enriched in dimorphic Ajellomycetaceae but not in saprophytic species was protein kinase activity (Table S2), we further classified protein kinases using Kinannote32, including the divergent fungal-specific protein kinase (FunK1; Methods). We found an expansion of the FunK1 family in dimorphic Ajellomycetaceae as previously reported5,14. As we increased the phylogenetic density in the Ajellomycetaceae, including species and strains with high genetic and phenotypic variation, this allowed resolution of the evolutionary history of this family of kinases (Fig. 5). The FunK1 family is enriched in the systemic pathogens B. dermatitidis/gilchristii, H. capsulatum, Paracoccidioides, but limited in saprophytic species, H. griseus and P. hystricis. Notably, this family is also limited in the adiaspore-forming Ea. parva (UAMH130, UAMH139), Ea. crescens (UAMH4076) but not in Ea. crescens (UAMH3008). In addition, we found that the recently described primary pathogen B. percursus includes a large expansion of FunK1 kinases, with almost twice the number as compared with its closest relative B. dermatitidis (Fig. 5). Phylogenetic analysis of FunK1 genes revealed that this family undergone independent lineage-specific expansions in primary dimorphic pathogens of both Ajellomycetaceae and Onygenaceae, but not in non-pathogenic or opportunistic dimorphic species (Fig. 5).

Expansion and contraction of protein kinases class FunK1 across the Ajellomycetaceae. (top) FunK1 A and FunK1 B counts for each taxon across the Ajellomycetaceae species and other compared genomes. The tree shown corresponds to the maximum likelihood phylogenetic tree of the family Ajellomycetaceae from Fig. 1. (bottom) Maximum likelihood tree of FunK1 family showing expansion in Ajellomycetaceae yeast-forming (blue) pathogenic species. Each species (top) or gene (bottom) has a color code indicating the parasitic phase at 37 °C (yeast, adiaspore, spherule, or non-pathogenic).
To identify specific genes that evolved within the Ajellomycetaceae to enable the dimorphic transition and infection of mammals, we identified and examined ortholog clusters that were unique to these pathogens and absent in both H. griseus and P. hystricis. We identified 75 ortholog clusters that were present in dimorphic Ajellomycetaceae and dimorphic Onygenaceae species but absent in H. griseus and P. hystricis (i.e. ‘sporophyte gene loss events’), and 212 ortholog clusters that were present in dimorphic Ajellomycetaceae, but absent in the dimorphic Onygenaceae species, H. griseus and P. hystricis (i.e. ‘dimorphic Ajellomycetaceae gene gain events’; Table S5). The gene loss events include a mold specific protein (CIMG_00805), a phenol 2-monooxygenase (BDFG_05966), as well as several protein kinases and transporters. The gene gain events include several protein kinases, transporters, transcription factors, and peptidases (Table S5). In addition, we annotated genes that have been broadly associated with host-pathogen interactions (Pathogen Host interaction database PHI), and genes that have been reported as virulence factors in fungal species (Database of Fungal Virulence Factors DFVF)33,34. We found that many genes that have been associated with host-pathogen interactions are present in the saprophytic P. hystricis and H. griseus, and in fact some appear at higher copy number counts (Table S5). This emphasizes that host-pathogen interaction and virulence factors are not only important for pathogenicity in mammals, but also for saprophytic species to survive and adapt to stress in the environment, including for example the ability to evade the engulfment by amoebae, or the interaction with insects35,36.
Conservation of genes induced during in vivo infection
We also hypothesized that genes induced during mammalian infection in dimorphic Ajellomycetaceae pathogens might have different gene conservation patterns among Blastomyces, Histoplasma, Paracoccidioides, and the novel species and saprophytes. Transcriptional responses during an in vivo mouse model of infection had been studied in B. dermatitidis (strain ATCC26189;5) and P. brasiliensis (strain Pb18;37). In both B. dermatitidis and P. brasiliensis the high-affinity zinc transporter, ZRT1 (BDFG_09159; PADG_06417) and the flavodoxin-like protein PST2 (BDFG_08006; PADG_07749) were highly induced during infection; these genes are conserved in all Ajellomycetaceae included in this analysis (Table S6). Of the 72 in vivo-induced genes in B. dermatitidis ATCC26188, 22% are absent from the P. hystricis genome and 11% are absent from the H. griseus genome. The genes absent in P. hystricis include the cysteine dioxygenase (BDFG_08059), an acetyltransferase (BDFG_07903), a thioesterase (BDFG_02348), a superoxide dismutase (BDFG_07895), a sodium/hydrogen exchanger (BDFG_05427), and a response regulator protein (BDFG_01066). In addition, we identified three secreted proteins that were found induced during the interaction of B. dermatitidis with macrophages (BDFG_08689, BDFG_06057, BDFG_03876)5 that are found conserved only in the dimorphic Ajellomycetaceae pathogens but not in H. griseus, P. hystricis, or outgroup species. These are strong candidates for effector proteins involved in host infection. Similar numbers of genes found induced during in vivo infection in P. brasiliensis were absent in P. hystricis and H. griseus, (23% and 17.5%, respectively; Table S6). Genes absent in both species include Gpr1 family protein (PADG_08695), isovaleryl-CoA dehydrogenase (PADG_05046), glutathione-dependent formaldehyde-activating (PADG_05832), predicted transmembrane protein (PADG_05761), and ergot alkaloid biosynthetic protein A (PADG_07739). Many of the genes that were absent in P. hystricis and H. griseus, were also absent in at least one of the dimorphic pathogens in the Ajellomycetaceae, highlighting species-specific gene gain events that may confer virulence specialization.
Evolution of the mating type locus in the Onygenales family Ajellomycetaceae
To elucidate the evolution of the mating system in the Ajellomycetaceae, we identified and characterized the mating locus of P. hystricis, and H. griseus. Notably, these two species have both mating type idiomorphs HMG box (MAT 1-2) and alpha box (MAT1-1); these are therefore the only homothallic species identified so far within the Ajellomycetaceae (Fig. 6). Comparative analysis showed that the locus is not expanded relative to other Ajellomycetaceae species (21 kb between the flanking genes SLA2 to APN2/COX13), unlike the expansion that observed in the larger genomes of B. dermatitidis or B. gilchristii (~60 kb;38). One difference is that a sequence inversion between APN2 and COX13 is uniquely observed in P. hystricis relative to all other sequenced Ajellomycetaceae species, including H. griseus and previously reported species5,15,38,39. This inversion observed in P. hystricis (mating type idiomorph flanked by SLA2 and APN2/COX13) is similar to the gene order in the Onygenaceae species Coccidioides spp. but not in the Arthrodermataceae species M. gypseum (Nannizzia gypsea40) or T. rubrum (Fig. 6; Table S7), suggesting independent inversion events within the locus. In addition, we identified the mating type locus in non-pathogenic Onygenaceae, and found that Amauroascus mutatus and Byssoonygena ceratinophila have both mating type idiomorphs (Table S7). In A. mutatus, HMG box (MAT 1-2) and alpha box (MAT1-1) are linked in the same scaffold, however they are separated by 184 kb, including 65 protein-coding genes (Fig. 6). The identification of homothallic mating loci in non-pathogenic species within both the Ajellomycetaceae and the Onygenaceae suggests that there have been multiple transitions between homothallic and heterothallic sexual states in dimorphic pathogens and non-pathogenic species in the Onygenales.

Mating type evolution within the Onygenales family Ajellomycetaceae. Synteny schema depicting orientation and conservation of the genes adjacent to the mating type locus idiomorphs HMG box (MAT 1-2, red) and alpha box (MAT1-1, dark red) in the Ajellomycetaceae. The more basal species, P. hystricis, has both mating type idiomorphs intact, being the only homothallic species identified so far within the Ajellomycetaceae. The locus has similar size to other Ajellomycetaceae species with 21 kb average from SLA2 (blue) to APN2/COX13 (green/orange), but not Blastomyces dermatitidis/gilchristii (60 kb; Li W et al.38). A synteny inversion encompassing APN2 and COX13 is uniquely observed in this ancestor relative to the rest of Ajellomycetaceae species included in this study, including H. griseus. Grey genes are mating locus hypothetical proteins (MLHP).
Discussion
The evolution of the dimorphic Onygenales is characterized by multiple transitions between saprophytic and human pathogenic growth. By sequencing the genomes of two additional dimorphic adiaspore-forming species (Emmonsia parva, Ea. crescens) and two early diverging non-pathogenic, non-dimorphic species (Helicocarpus griseus and Polytolypa hystricis), we analyzed how changes in gene content correlate with transitions to pathogenesis. Our results establish an outer bound on the timing of two key events in the evolution of the Ajellomycetaceae; both the loss of genes involved in digesting plant cell walls and the expansion of proteases, transcriptional regulators, and protein kinases, occurred after divergence with H. griseus. The shift in these families was previously observed in both Paracoccidioides within this group and in the related Coccidioides–Uncinocarpus clade14,17; more recently the comparison with non-pathogenic Onygenaceae species supported previous reports of the contraction of gene families involved in digesting plant cell walls throughout the Onygenales, and of the expansion of gene families involved in digesting animal protein20. Whereas the studies on contraction of gene families involved in digesting plant cell walls addressed other fungi outside the order Onygenales, our analysis shows that H. griseus and P. hystricis did not experience such a contraction, which suggests that contractions in gene families involved in digesting plant cell walls throughout the Onygenales have occurred multiple times independently and more recently in Onygenaceae and Ajellomycetaceae families.
Since many virulence factors and dimorphic switch related proteins were also conserved in the rarely pathogenic (Ea. parva and Ea. crescens) and in the non-dimorphic Ajellomycetaceae species (P. hystricis and H. griseus), this suggests that these proteins could be needed not only for survival in the host, and that presence of virulence factors alone does not specify pathogenicity. In dimorphic Ajellomycetaceae other mechanisms such as gene regulation or selection may play a role in how these genes respond during distinct stimuli. In addition, it is possible that those virulence factors in mammals were also needed for the survival in the soil, for example to survive interaction with amoebae or in insects41. However, there are some virulence factors that are needed for virulence in mammals that are not necessary for virulence in other eukaryotes. For example, the alpha mating factor locus in Cryptococcus neoformans contributes to virulence in mice, but no difference was observed in the interaction of congenic MATα and MATa strains with amoebae42. While the human host is a very different habitat in comparison with soil or excrements of animals that are the most common natural niche for this group of fungi, interactions with other eukaryotes in the environment likely selected for some properties that also predisposed species to become pathogenic to humans.
Selection pressures in the environment are responsible for the emergence and maintenance of traits that confer upon some soil microbes the capacity for survival in animal hosts. This is important to understand the evolution and emergence of novel pathogens. For example, Emergomyces africanus has recently emerged and numerous cases of novel yeast-forming species have been reported. However, Emergomyces species have not been found in other mammals like rodents or in the soil. In addition, Es. africanus has a very restricted geographic distribution. Across the Onygenales, nearly all species have a high lower ratio of proteases to carbohydrate active enzymes; the exceptions are the two saprophytes P. hystricis and H. griseus, but not the saprophytes related to Coccidioides such as Uncinocarpus reesii. This genomic profile matches the results of growth assays for the non-pathogenic species U. reesii; this species can grow on a wide range of proteinaceous substrates but only a very limited range of carbohydrates, namely cellulose and its component glucose14. Together, these findings suggest that fungi in the Onygenales transitioned from saprophytes that digest plant materials to saprophytes that digest more limited plant materials and a wide range of animal proteins, and finally those that are major animal pathogens.
We found many of the genes with roles in the dimorphic transition and virulence are conserved in Ea. parva and Ea. crescens, and in saprophytic species H. griseus and P. hystricis, and we did not find larger patterns of gain of functional classes in pathogenic species. However, we found smaller changes in gene content related to the regulation of gene expression and signaling that may account for the ability to grow at high temperature, species specific morphological transitions, and therefore their differences in virulence, since rapid thermo-tolerance and morphological adaptation is essential for pathogenesis29,30,43. Overall, we found significant expansions of the number of the protein kinase FunK1 family, transcription factors and other genes associated with the regulation of gene expression in yeast-forming Ajellomycetaceae relative to adiaspore-forming and saprophytic species. Our comparison with saprophytic Ajellomycetaceae highlights dramatic decreases and complete absences of several classes of gene families that are important for survival and growth in a soil environment.
Intriguingly, we found that both more basal non-pathogenic Ajellomycetaceae species H. griseus and P. hystricis appear homothallic, in striking contrast to all the previously described largely pathogenic species in this group that are all heterothallic. P. hystricis has a fused mating type locus, with both idiomorphs, HMG box (MAT 1-2) and alpha box (MAT1-1), fused at a single locus, and the same is likely true of H. griseus, as genes are located at the ends of two scaffolds in this assembly. In addition, we found that this is also true for the non-pathogenic Onygenaceae species Amauroascus mutatus and Byssoonygena ceratinophila; these species have both mating type idiomorphs. A. mutatus has both mating type idiomorphs located on the same scaffold; however, these are separated by a much larger distance than is typically observed at the MAT locus of homothallic fungi. This suggests that this locus has been subject to recombination events either to bring the two idiomorphs nearby on the same scaffold or alternatively in expansion of a fused locus. While it has been previously suggested that the ancestor of the Onygenales very likely was heterothallic39, the placement of multiple lineages of homothallic species in this group raises the possibility of a homothallic ancestor, with more recent transitions in pathogenic species to be heterothallic and rearrangements of the mating locus in others such as P. hystricis and A. mutatus. Both P. hystricis UAMH7299 and H. griseus UAMH5409 can develop ascomata and ascospores, which have been shown to sporulate in P. hystricis11,27,44. Loss of the capacity for homothallic self-mating could increase the frequency of out-crossing and this may be beneficial to some species under pressure from their environment or host45 such as the pathogenic species in this group. Examining the mating type loci of additional non-pathogenic species in the Onygenales would further reveal if there are novel configurations of homothallic or heterothallic loci, which may help infer the mating ability of ancestral species and the timing of transitions in pathogens and non-pathogens.
Our phylogenomic analysis revealed that dimorphic pathogens in the Onygenales have undergone multiple evolutionary transitions that enabled infection of humans and other mammals. Some such as the loss of plant cell wall degrading enzymes are convergent for pathogens in the two families of Onygenales. Our comparisons also revealed major differences in non-pathogenic species between these two families, which suggested that some species appear intermediate between saprophytes and pathogens in terms of having a shift in metabolic capacity without associated pathogenic growth. Targeting additional intermediate species within the Ajellomycetaceae with different phenotypes (e.g. Emmonsiellopsis) for genome sequencing and comparison would help further refine the picture of the overall evolution of this important group of fungi.
Materials and Methods
Genome sequencing
Species were selected for sequencing based on previous estimates of high genetic variation among the members of the genus Emmonsia, especially in the Ea. parva species, and based on observations of the emergence of Emmonsia-like species in the Ajellomycetaceae causing systemic human mycoses worldwide8,9. We selected one strain of Emmonsia parva (UAMH130; CBS139881; type strain) and one additional strain of Emmonsia crescens (UAMH4076; CBS139868). Ea. parva UAMH130 is from the lungs of a rodent in the USA, and Ea. crescens UAMH4076 is from a greenhouse source in Canada, and mated with Ea. crescens UAMH129, 34922. In addition, the first strictly non-pathogenic non-dimorphic species from the Ajellomycetaceae, one strain of Helicocarpus griseus (UAMH5409; a.k.a. Spiromastix grisea) and Polytolypa hystricis (UAMH7299) were selected for whole genome sequencing. Genomic DNA was isolated from mycelial phase at 22 °C. We constructed one library with 180-base inserts and sequenced on the Illumina HiSeq 2000 platform to generate 101 bp paired-end reads with high coverage (173× UAMH4076; 199× UAMH130; 205× UAMH5409; 163× UAMH7299).
Genome assembly, gene prediction and annotation
The 101-bp Illumina reads of Ea. parva UAMH130, Ea. crescens UAMH4076, P. hystricis and H. griseus were assembled using ALLPATHS-LG46 with default parameters. All four de novo assemblies were evaluated using the GAEMR package (http://software.broadinstitute.org/software/gaemr/), which revealed no aberrant regions of coverage, GC content or contigs with sequence similarity suggestive of contamination. Scaffolds representing the mitochondrial genome were separated out from the nuclear assembly.
Genes were predicted and annotated by combining calls from multiple methods to obtain the best consensus model for a given locus. These included ab initio predictions (GlimmerHMM, Augustus, Snap, GeneMark-ES), homologous inference (Genewise, TBlastN), and gene model consolidation programs (EvidenceModeler)47,48. For the protein coding-gene name assignment we combined HMMER PFAM/TIGRFAM, Swissprot and Kegg products. Kinannote was used to annotate protein kinases32. To evaluate the completeness of predicted gene sets, the representation of core eukaryotic genes was analyzed using CEGMA genes23 and BUSCO24.
Identification of orthologs and phylogenomic analysis
To compare gene content and conservation, we identified orthologous gene clusters in these sequenced genomes and in other Onygenales genomes using OrthoMCL (version 1.4) with a Markov inflation index of 1.5 and a maximum e-value of 1e-549. We included the genomes of the classical dimorphic pathogenic species (Blastomyces dermatitidis, B. gilchristii, Histoplasma capsulatum, Paracoccidioides brasiliensis, P. lutzii, Coccidioides immitis, and C. posadasii), novel dimorphic human pathogenic species (Blastomyces percursus, Emergomyces africanus, Es. pasteurianus), and two dimorphic non-human pathogenic species Emmonsia parva UAMH139 and Emmonsia crescens, along with other species, including three Aspergillus (Table S1; Figure S7). In addition, we annotated and included in the compare gene content and conservation analysis the available genomes of saprophytic species from the family Onygenaceae Byssoonygena ceratinophila (UAMH5669), Amauroascus mutatus (UAMH3576), Amauroascus niger (UAMH3544), and Chrysosporium queenslandicum (CBS280.77), and the genome of the yeast-forming dimorphic pathogen Emergomyces orientalis (5Z489, Table S1). Although, this is the largest genomic data set of Ajellomycetaceae strains to date, the number of shared gene families after each addition of genome data did not clearly reach a plateau; this suggests that the current estimate of a core of 4,200 genes is too high, and that more genomes are needed to define the core-genome and pan-genome in the Ajellomycetaceae (Figure S6).
To examine the phylogenetic relationship of the newly sequenced Ea. parva and Ea. crescens, P. hystricis and H. griseus relative to other dimorphic fungi we used single copy orthologs determined and clustered using OrthoMCL. A total of 31 genomes from the Onygenales order and three Aspergillus genomes were chosen to estimate the species phylogeny (Table S1). These include the four newly strains described here: Ea. parva UAMH130, Ea. crescens UAMH4076, P. hystricis UAMH7299 and H. griseus UAMH5409, as well as the following: three Blastomyces dermatitidis (ATCC26199, ATCC18188, ER-3), one Blastomyces gilchristii (SLH14081), one Emmonsia parva (UAMH139), one Emmonsia crescens (UAMH3008), two Histoplasma (WU24, G186AR), five Paracoccidioides (Pb01, Pb03, Pb18, PbCnh, Pb300), two Coccidioides (RS, Silveira), Uncinocarpus reesii (UAMH1704), Microsporum gypseum (CBS118893), Trichophyton rubrum (CBS118892), Aspergillus nidulans (FGSC A4), A. flavus (NRRL3357) and A. fumigatus (Af293). Protein sequences were aligned using MUSCLE, and a phylogeny was estimated from the concatenated alignments using RAxML v7.7.850 with model PROTCATWAG with a total of 1,000 bootstrap replicates.
Gene family and protein domain analysis
To identify gene content changes that could play a role in the evolution of the dimorphism and pathogenesis within Ajellomycetaceae, we searched for expansions or contractions in functionally classified genes compared to the other fungi from the order Onygenales. Genes were functionally annotated by assigning PFAM domains, GO terms, and KEGG classification. HMMER351 was used to identify PFAM domains using release 27. GO terms were assigned using Blast2GO52, with a minimum e-value of 1 × 10−10. Protein kinases were identified using Kinannote32 and divergent FunK1 kinases were further identified using HMMER3. We further annotated carbohydrate active enzymes, peptidases, and transporter families using the CAZY version 07-15-201653, MEROPS version 9.1254, and TCDB version 01-05-201728 databases, respectively. Proteins were searched against their corresponding databases using BLAST, with minimum e-values of 1 × 10−80 for CAZY, 1 × 10−20 for MEROPS, and 1 × 10−20 for TCDB.
To identify functional enrichments in the compared genomes, we used four gene classifications: OrthoMCL similarity clusters, PFAM domains, KEGG pathways, and Gene Ontology (GO) including different hierarchy levels, MEROPS and CAZY categories, and transporter families. Using a matrix of gene class counts for each classification type, we identified enrichment comparing two subsets of queried genomes using Fisher’s exact test. Fisher’s exact test was used to detect enrichment of PFAM, KEGG, GO terms, CAZY, MEROPS, and transporter families between groups of interest, and p-values were corrected for multiple comparisons55. Significant (corrected p-value < 0.05) gene class expansions or depletions were examined for different comparisons.
Data Availability Statement
The raw sequences, genome assemblies and gene annotations were deposited in GenBank under the following BioProject accession numbers: Emmonsia parva strain UAMH130 (PRJNA252752), Emmonsia crescens strain UAMH4076 (PRJNA252751), Polytolypa hystricis strain UAMH7299 (PRJNA234736), and Helicocarpus griseus strain UAMH5409 (PRJNA234735).
References
Klein, B. S. & Tebbets, B. Dimorphism and virulence in fungi. Curr Opin Microbiol 10, 314–9 (2007).
Sil, A. & Andrianopoulos, A. Thermally dimorphic human fungal pathogens–Polyphyletic pathogens with a convergent pathogenicity trait. Cold Spring Harb Perspect Med 5, a019794 (2015).
Gauthier, G. M. Dimorphism in fungal pathogens of mammals, plants, and insects. PLoS Pathog 11, e1004608 (2015).
England, D. M. & Hochholzer, L. Adiaspiromycosis: an unusual fungal infection of the lung. Report of 11 cases. Am J Surg Pathol 17, 876–86 (1993).
Munoz, J. F. et al. The dynamic genome and transcriptome of the human fungal pathogen Blastomyces and close relative Emmonsia. PLoS Genet 11, e1005493 (2015).
Herr, R. A. et al. Phylogenetic analysis of Lacazia loboi places this previously uncharacterized pathogen within the dimorphic Onygenales. J Clin Microbiol 39, 309–14 (2001).
Kenyon, C. et al. A dimorphic fungus causing disseminated infection in South Africa. N Engl J Med 369, 1416–1424 (2013).
Schwartz, I. S. et al. 50 Years of Emmonsia disease in humans: the dramatic emergence of a cluster of novel fungal pathogens. PLoS Pathog 11, e1005198 (2015).
Dukik, K. et al. Novel taxa of thermally dimorphic systemic pathogens in the Ajellomycetaceae (Onygenales). Mycoses 60, 296–309 (2017).
Wang, O. et al. A novel dimorphic pathogen, Emergomyces orientalis (Onygenales), agent of disseminated infection. Mycoses (in press), (2017).
Marin-Felix, Y. et al. Emmonsiellopsis, a new genus related to the thermally dimorphic fungi of the family Ajellomycetaceae. Mycoses 58, 451–60 (2015).
Untereiner, W. A. et al. The Ajellomycetaceae, a new family of vertebrate-associated Onygenales. Mycologia 96, 812–821 (2004).
Untereiner, W. A., Scott, J. A., Naveau, F. A., Currah, R. S. & Bachewich, J. Phylogeny of Ajellomyces, Polytolypa and Spiromastix (Onygenaceae) inferred from rDNA sequence and non-molecular data. Stud Mycol 47, 25–35 (2002).
Desjardins, C. A. et al. Comparative genomic analysis of human fungal pathogens causing paracoccidioidomycosis. PLoS Genet 7, e1002345 (2011).
Munoz, J. F. et al. Genome diversity, recombination, and virulence across the major lineages of Paracoccidioides. mSphere 1 (2016).
Neafsey, D. E. et al. Population genomic sequencing of Coccidioides fungi reveals recent hybridization and transposon control. Genome Res 20, 938–46 (2010).
Sharpton, T. J. et al. Comparative genomic analyses of the human fungal pathogens Coccidioides and their relatives. Genome Res 19, 1722–31 (2009).
Burmester, A. et al. Comparative and functional genomics provide insights into the pathogenicity of dermatophytic fungi. Genome Biol 12, R7 (2011).
Martinez, D. A. et al. Comparative genome analysis of Trichophyton rubrum and related dermatophytes reveals candidate genes involved in infection. MBio 3, e00259–12 (2012).
Whiston, E. & Taylor, J. W. Comparative phylogenomics of pathogenic and nonpathogenic species. G3 Bethesda 6, 235–44 (2015).
Munoz, J. F. et al. Genome update of the dimorphic human pathogenic fungi causing paracoccidioidomycosis. PLoS Negl Trop Dis 8, e3348 (2014).
Sigler, L. Ajellomyces crescens sp. nov., taxonomy of Emmonsia spp., and relatedness with Blastomyces dermatitidis (teleomorph Ajellomyces dermatitidis). J Med Vet Mycol 34, 303–14 (1996).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–7 (2007).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Peterson, S. W. & Sigler, L. Molecular genetic variation in Emmonsia crescens and Emmonsia parva, etiologic agents of adiaspiromycosis, and their phylogenetic relationship to Blastomyces dermatitidis (Ajellomyces dermatitidis) and other systemic fungal pathogens. J Clin Microbiol 36, 2918–25 (1998).
Blin, K. et al. antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 45, W36–W41 (2017).
Gamble, W. R., Gloer, J. B., Scott, J. A. & Malloch, D. Polytolypin, a new antifungal triterpenoid from the coprophilous fungus Polytolypa hystricis. J. Nat. Prod. 58, 1983–1986 (1995).
Saier, M. H. Jr., Tran, C. V. & Barabote, R. D. TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res 34, D181–6 (2006).
Beyhan, S., Gutierrez, M., Voorhies, M. & Sil, A. A temperature-responsive network links cell shape and virulence traits in a primary fungal pathogen. PLoS Biol 11, e1001614 (2013).
Nemecek, J. C., Wuthrich, M. & Klein, B. S. Global control of dimorphism and virulence in fungi. Science 312, 583–8 (2006).
Rappleye, C. A., Eissenberg, L. G. & Goldman, W. E. Histoplasma capsulatum alpha-(1,3)-glucan blocks innate immune recognition by the beta-glucan receptor. Proc Natl Acad Sci USA 104, 1366–1370 (2007).
Goldberg, J. M. et al. Kinannote, a computer program to identify and classify members of the eukaryotic protein kinase superfamily. Bioinformatics 29, 2387–94 (2013).
Lu, T., Yao, B. & Zhang, C. DFVF: database of fungal virulence factors. Database J. Biol. Databases Curation 2012 (2012).
Winnenburg, R. et al. PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 34, D459–D464 (2006).
Casadevall, A., Steenbergen, J. N. & Nosanchuk, J. D. ‘Ready made’ virulence and ‘dual use’ virulence factors in pathogenic environmental fungi-the Cryptococcus neoformans paradigm. Curr Opin Microbiol 6, 332–337 (2003).
Derengowski Lda, S. et al. The transcriptional response of Cryptococcus neoformans to ingestion by Acanthamoeba castellanii and macrophages provides insights into the evolutionary adaptation to the mammalian host. Eukaryot Cell 12, 761–74 (2013).
Pigosso, L. L. et al. Paracoccidioides brasiliensis presents metabolic reprogramming and secretes a serine proteinase during murine infection. Virulence 0, 1–18 (2017).
Li, W. et al. Identification of the mating-type (MAT) locus that controls sexual reproduction of Blastomyces dermatitidis. Eukaryot Cell 12, 109–17 (2013).
Li, W., Metin, B., White, T. C. & Heitman, J. Organization and evolutionary trajectory of the mating type (MAT) locus in dermatophyte and dimorphic fungal pathogens. Eukaryot Cell 9, 46–58 (2010).
Hoog, G. Sde et al. Toward a novel multilocus phylogenetic taxonomy for the dermatophytes. Mycopathologia 182, 5–31 (2017).
Steenbergen, J. N., Shuman, H. A. & Casadevall, A. Cryptococcus neoformans interactions with amoebae suggest an explanation for its virulence and intracellular pathogenic strategy in macrophages. Proc. Natl. Acad. Sci. 98, 15245–15250 (2001).
Nielsen, K. et al. Cryptococcus neoformans α strains preferentially disseminate to the central nervous system during coinfection. Infect. Immun. 73, 4922–4933 (2005).
Rappleye, C. A. & Goldman, W. E. Defining virulence genes in the dimorphic fungi. Annu Rev Microbiol 60, 281–303 (2006).
Sugiyama, M., Ohara, A. & Mikawa, T. Molecular phylogeny of onygenalean fungi based on small subunit ribosomal DNA (SSU rDNA) sequences. Mycoscience 40, 251–258 (1999).
Heitman, J., Sun, S. & James, T. Y. Evolution of fungal sexual reproduction. Mycologia 105, 1–27 (2013).
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci UA 108, 1513–8 (2011).
Haas, B. J., Zeng, Q., Pearson, M. D., Cuomo, C. A. & Wortman, J. R. Approaches to Fungal GenomeAnnotation. Mycology 2, 118–141 (2011).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13, 2178–89 (2003).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–90 (2006).
Eddy, S. R. Accelerated Profile HMM Searches. PLoS Comput Biol 7, e1002195 (2011).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–6 (2005).
Cantarel, B. L. et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37, D233–8 (2009).
Rawlings, N. D., Barrett, A. J. & Bateman, A. MEROPS: the peptidase database. Nucleic Acids Res 38, D227–33 (2010).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc Natl Acad Sci U A 100, 9440–5 (2003).
Acknowledgements
We thank Lynne Sigler for her help in selecting strains for sequencing. We thank the Broad Genomics Platform for generating Illumina sequence for this study, Sarah Young and Margaret Priest for their assistance in assembly and annotation, Christopher Desjardins for helpful comments on the manuscript, and Leslie Gaffney for help with Fig. 1. This project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under award N°: U19AI110818. This work was partly supported by Colciencias via the grants under Contract N°: 122256934875 and N°: 221365842971, and by the Universidad de Antioquia via a “Sostenibilidad 2016” grant.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: J.F.M., J.G.M., O.K.C., C.A.C. Performed the assembly and annotation: J.F.M., C.A.C. Analyzed the data: J.F.M., C.A.C. Contributed reagents/materials/analysis tools: J.G.M., O.K.C. Wrote the paper: J.F.M., C.A.C.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muñoz, J.F., McEwen, J.G., Clay, O.K. et al. Genome analysis reveals evolutionary mechanisms of adaptation in systemic dimorphic fungi. Sci Rep 8, 4473 (2018). https://doi.org/10.1038/s41598-018-22816-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22816-6
This article is cited by
-
Evolution of the human pathogenic lifestyle in fungi
Nature Microbiology (2022)
-
Phylogenetic and ecological reevaluation of the order Onygenales
Fungal Diversity (2022)
-
PRP8 Intein in Onygenales: Distribution and Phylogenetic Aspects
Mycopathologia (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
