
Abstract
Voltage-gated sodium channels NaV1.7, NaV1.8 and NaV1.9 have been the focus for pain studies because their mutations are associated with human pain disorders, but the role of NaV1.6 in pain is less understood. In this study, we selectively knocked out NaV1.6 in dorsal root ganglion (DRG) neurons, using NaV1.8-Cre directed or adeno-associated virus (AAV)-Cre mediated approaches, and examined the specific contribution of NaV1.6 to the tetrodotoxin-sensitive (TTX-S) current in these neurons and its role in neuropathic pain. We report here that NaV1.6 contributes up to 60% of the TTX-S current in large, and 34% in small DRG neurons. We also show NaV1.6 accumulates at nodes of Ranvier within the neuroma following spared nerve injury (SNI). Although NaV1.8-Cre driven NaV1.6 knockout does not alter acute, inflammatory or neuropathic pain behaviors, AAV-Cre mediated NaV1.6 knockout in adult mice partially attenuates SNI-induced mechanical allodynia. Additionally, AAV-Cre mediated NaV1.6 knockout, mostly in large DRG neurons, significantly attenuates excitability of these neurons after SNI and reduces NaV1.6 accumulation at nodes of Ranvier at the neuroma. Together, NaV1.6 in NaV1.8-positive neurons does not influence pain thresholds under normal or pathological conditions, but NaV1.6 in large NaV1.8-negative DRG neurons plays an important role in neuropathic pain.
Similar content being viewed by others


Introduction
Adult primary sensory neurons express five voltage-gated sodium channel isoforms (NaV), including TTX-sensitive (TTX-S) NaV1.1, NaV1.6 and NaV1.7, and TTX-resistant (TTX-R) NaV1.8 and NaV1.9 channels1. NaV1.7, NaV1.8 and NaV1.9, which are expressed preferentially in these neurons2,3,4, play critical roles in acute nociception and persistent pain in rodent models5,6,7,8,9. Identification and characterization of mutations in NaV1.7, NaV1.8 and NaV1.9 have confirmed causative links of these channels to human pain disorders10,11,12,13,14,15,16,17.
By contrast, NaV1.1 and NaV1.6, the two NaV subtypes widely expressed throughout the PNS and CNS, have been linked to CNS disorders such as epilepsy18,19,20,21. New evidence, suggests that their roles in nociception may have been underestimated. Activation of NaV1.1 using a spider toxin has been shown to produce robust pain behavior and profound hypersensitivity to mechanical stimuli, establishing the first evidence linking NaV1.1 to acute pain and mechanical allodynia22. NaV1.6 has been implicated in oxaliplatin-induced hyper-excitability in A-fiber neurons23 and cold allodynia24. Local knockdown of NaV1.6 using siRNA has also been shown to reduce spontaneous neuronal activity and nociceptive behaviors in rodent models of chronic pain25,26. Recently, the first NaV1.6 gain-of-function mutation associated with a human neuropathic pain disorder, trigeminal neuralgia, was reported27, highlighting the need to further investigate the role of NaV1.6 in the pain pathway.
A challenge to the elucidation of the mechanistic basis for the role of this channel in pain in animal models is that global NaV1.6 null mice are juvenile lethal28. To examine the specific contribution of NaV1.6 to the excitability of DRG neurons as well as acute and neuropathic pain behaviors, we have established two conditional NaV1.6 knockout models with NaV1.8-directed or AAV-mediated Cre-Lox recombination systems, to selectively knockout NaV1.6 in primary sensory neurons. Our data show that NaV1.6 contributes a substantial fraction of the TTX-S current in both small and large DRG neurons and manifests a long half-life at nodes of Ranvier in vivo, and that although NaV1.6 has limited role in acute nociception, it contributes to the development and maintenance of neuropathic pain behavior following spared nerve injury (SNI).
Results
Contribution of NaV1.6 to TTX-sensitive current in DRG neurons
The TTX-S sodium current activates during the initial depolarization phase and is important in setting the firing threshold of action potentials in DRG neurons29,30. We first examined the functional consequences of NaV1.6 deletion in Nav1.8-positive neurons by comparing TTX-S current in both small (20–25 µm) and large (40–45 µm) native DRG neurons from NaV1.6-heterozygous control and NaV1.8-driven NaV1.6-knockout (KO) mice. The conditional NaV1.6 knockout mice carry the Cre recombinase which is expressed at the native NaV1.8 locus31 and NaV1.6 alleles flanked with LoxP sites (NaV1.6flox/flox)32 as well as a Cre-dependent reporter cassette (loxP-stop-loxP-tdTomato fluorescent protein). This approach allows the visual identification (red fluorescence) of functional Cre recombinase expression in NaV1.8-positive neurons33 and thus provides a marker for the deletion of the floxed NaV1.6 alleles.
Representative traces of TTX-S currents recorded in red fluorescent small DRG neurons from Nav1.6-heterozygous control (Control: NaV1.6+/flox/NaV1.8+/Cre) and NaV1.8-driven NaV1.6 KO mice (NaV1.6Nav1.8 KO: NaV1.6flox/flox/NaV1.8+/Cre) showed a reduction of TTX-S current amplitude after NaV1.6 deletion (Fig. 1a). Peak TTX-S current density was calculated from the complete voltage-current relationship (Fig. 1b). Knocking out NaV1.6 resulted in ~34% reduction of the TTX-S current density in small DRG neurons (Control: 468 ± 59 pA/pF, n = 13; NaV1.6Nav1.8 KO 308 ± 50 pA/pF, n = 15; p = 0.0253 unpaired t test test) (Fig. 1c). By contrast, the TTX-R current density in these neurons was not affected by NaV1.6 deletion (Control: 59.6 ± 6.6 pA/pF, n = 12; NaV1.6Nav1.8 KO: 52.1 ± 5.3 pA/pF, n = 14).

Contribution of NaV1.6 to TTX-sensitive sodium current in small and large DRG neurons in NaV1.8 driven Nav1.6 knock out mice. (a) Representative traces of TTX-S currents recorded in small DRG neurons from heterozygous NaV1.8+/CreNaV1.6+/flox (Control, black lines) and NaV1.8+/CreNaV1.6flox/flox (NaV1.6Nav1.8 KO, orange lines) mice. Cells were held at −100 mV and stepped to membrane potentials from −80 to +30 mV in 5 mV increments (100 ms) to record voltage-dependent sodium currents. (b) The current-voltage (I-V) relation curves for TTX-S sodium currents in small Control (n = 13) and NaV1.6Nav1.8 KO (n = 15) KO DRG neurons. (c) Current densities of TTX-S sodium currents in small Control (n = 13) and NaV1.6Nav1.8 KO (n = 15) DRG neurons. (d) Representative traces of TTX-S sodium currents recorded in large DRG neurons from heterozygous NaV1.8+/CreNaV1.6+/flox (Control) and NaV1.8+/CreNav1.6flox/flox (NaV1.6Nav1.8 KO) mice. Cells were held at −70 mV, prepulsed to −100 mV for 500 ms, and stepped to membrane potentials from −65 mV to +40 mV in 5 mV increments (100 ms) to record the voltage-dependent sodium currents. (e) The current-voltage (I-V) relation curves for TTX-S sodium currents in large Control (n = 5) and NaV1.6Nav1.8 KO (n = 7) DRG neurons. (f) Current densities of TTX-S sodium currents in large Control (n = 5) and NaV1.6Nav1.8 KO (n = 7) DRG neurons. Data are presented as mean ± SEM. *p < 0.05 based on unpaired t test.
Representative traces of TTX-S currents recorded in large red fluorescent DRG neurons from control and NaV1.6Nav1.8 KO mice showed a reduction of TTX-S current amplitude after NaV1.6 deletion (Fig. 1d). As shown in Fig. 1e, the average TTX-S sodium currents in these neurons were substantially reduced in NaV1.6Nav1.8 KO mice as compared to the control heterozygous littermates. The density of TTX-S sodium currents in large DRG neurons lacking NaV1.6 channels was significantly reduced by 53% compared to control neurons (Control: 1557 ± 328 pA/pF, n = 5; NaV1.6Nav1.8 KO: 731 ± 157 pA/pF, n = 7; p = 0.0317) (Fig. 1f). In contrast, the current density of TTX-R sodium currents in these large DRG neurons (mostly produced by NaV1.8 channels) was similar between control and NaV1.6Nav1.8 KO mice (Control: 329 ± 48 pA/pF, n = 5; NaV1.6Nav1.8 KO, 334 ± 41 pA/pF, n = 7; p = 0.931). Our results suggest that approximately 80% of the voltage-dependent sodium currents in NaV1.8-positive large DRG neurons are TTX-S, and more than one half of the TTX-S current is carried by NaV1.6 sodium channels.
Although real-time RT-PCR did not show substantial reduction in NaV1.6 RNA in the conditional knockout mice (Supplemental Fig. 1), the presence of red fluorescence indicates the expression of functional Cre recombinase in NaV1.8-positive neurons, and the reduction in the TTX-S current is consistent with the knockout of NaV1.6 in these neurons.
Because NaV1.8 channels are only expressed in a subpopulation of large DRG neurons33, we performed voltage-clamp recordings in a global NaV1.6 KO mouse strain (MED: Scn8amedtg) and homozygous wild-type littermates (WT), which were genotyped prior to tissue harvesting, in order to assess the contribution of NaV1.6 to the TTX-S current in neurons that do not express NaV1.8. The full knockout of NaV1.6 in Scn8amedtg was verified at the RNA level using real-time RT-PCR (Supplemental Fig. 1) and at the protein level by the total loss of NaV1.6 staining at nodes of Ranvier in the sciatic nerve (Supplementa1 Fig. 2).
Representative traces of TTX-S currents recorded in large DRG neurons of WT and Scn8amedtg mice are shown in Fig. 2a and b, respectively. We recorded TTX-R sodium currents (mostly produced by NaV1.8 channels under the recording conditions in these experiments) in 20% of large DRG neurons (4 out of 20 cells) from Scn8amedtg mice and 26% (6 out of 23 cells) from wild-type littermates. In large neurons with TTX-R currents (Fig. 2c) and without TTX-R currents (Fig. 2d), the amplitude of TTX-S sodium currents was significantly reduced in Scn8amedtg mice, compared to their WT littermates. The average TTX-S current density was reduced by 47% in Scn8amedtg large DRG neurons that express TTX-R sodium currents (WT: 701 ± 98 pA/pF, n = 6; MED, 374 ± 80 pA/pF, n = 4; p = 0.045) (Fig. 2e). In large DRG neurons that do not express TTX-R sodium currents, the average TTX-S current density was reduced by 60% (WT: 764 ± 66 pA/pF, n = 17; MED: 304 ± 44 pA/pF, n = 16; p = 3.04E-6) (Fig. 2f). The current density of TTX-R sodium currents in large DRG neurons was unaffected in Scn8amedtg mice (WT: 197 ± 30 pA/pF, n = 6; MED: 156 ± 43 pA/pF, n = 4; p = 0.443) (Fig. 2g). In addition, the current density of TTX-S sodium currents for WT cells that express TTX-R sodium currents was not significantly different from cells that do not express TTX-R currents (p = 0.625). Our data from Scn8amedtg mice not only confirmed our findings in NaV1.8-driven NaV1.6 knockout mice, but also showed that NaV1.6 accounts for up to 60% of the TTX-S current in neurons that do not express TTX-R currents.

Contribution of NaV1.6 to TTX-sensitive sodium current in large DRG neurons in NaV1.6 global knockout (SCN8amedtg) mice. Representative traces of TTX-S currents recorded in large DRG neurons from (a) wild-type (WT) mice and (b) NaV1.6 global KO (SCN8amedtg, MED) mice. (c) The current-voltage (I-V) relationship curves for TTX-S sodium currents in large WT (n = 6) and MED (n = 4) DRG neurons that express TTX-R sodium currents. (d) The current-voltage (I-V) relationship curves for TTX-S sodium currents in large WT (n = 17) and MED (n = 16) DRG neurons that do not express TTX-R sodium currents. (e) Current densities of TTX-S sodium currents in large WT (n = 6) and MED (n = 4) DRG neurons that express TTX-R sodium currents. (f) Current densities of TTX-S sodium currents in large WT (n = 17) and MED (n = 16) DRG neurons that do not express TTX-R sodium currents. (g) Current densities of TTX-R sodium currents in large WT (n = 6) and MED (n = 4) DRG neurons. (h) Representative traces of resurgent sodium currents recorded in large DRG neurons that do not express TTX-R sodium currents from WT mice. (i) Representative traces show lack of resurgent sodium currents from a large DRG neurons that do not express TTX-R sodium currents from MED mice. Data are presented as mean ± SEM. *p < 0.05, ***p < 0.001 based on unpaired t test.
Nav1.6 has been shown to conduct resurgent currents in DRG neurons34. Representative traces for resurgent sodium currents from WT and Scn8amedtg mice are shown in Fig. 2h and i, respectively. The average amplitude of resurgent sodium currents in WT large neurons is 1.64 ± 0.17% of the peak current. We recorded resurgent sodium currents in 82% of WT large DRG neurons that do not express TTX-R sodium currents (14 out of 17 cells) while none of the cells (0 out of 16 cells) from Scn8amedtg mice produced resurgent currents.
NaV1.8-Cre driven NaV1.6 KO mice manifest normal acute and persistent pain behavior
The electrophysiological recordings in DRG neurons described above demonstrated that NaV1.6 deletion markedly reduced the TTX-S current in both small and large DRG neurons. Because global NaV1.6 is juvenile lethal, we examined whether pain behavior was altered in the NaV1.6Nav1.8 KO mice. Surprisingly, the NaV1.6Nav1.8 KO mice were phenotypically indistinguishable from their heterozygous control littermates in all behavioral tests we conducted. Conditional NaV1.6 knockout and control mice performed equally well in the rotarod test, suggesting that sensorimotor function was not affected (Fig. 3a). Withdrawal thresholds to mechanical stimuli assessed using von Frey and Randall Selitto tests were also not significantly different in NaV1.6Nav1.8 KO mice compared to their heterozygous control littermate (Fig. 3b,c). In addition, conditional NaV1.6 knockout and control mice displayed similar acute thermal pain thresholds in the Hargreaves test (radiant heat applied to hind paw) and the Hot Plate test (mice placed on a metal plate at 52 °C or 56 °C) (Fig. 3d,e). Thermal preference test was performed to examine whether deletion of NaV1.6 impacts avoidance behavior. Compared to control mice, no difference was observed in NaV1.6Nav1.8 KO mice at all the temperatures tested (non-noxious cool temperatures 20 °C or 28 °C, and non-noxious heat temperature 38 °C or 46 °C) (Fig. 3f).

Motor and acute pain behaviors in NaV1.8-Cre driven NaV1.6 KO mice. (a) Motor function: rotarod test (Control, n = 6; NaV1.6Nav1.8 KO, n = 9). (b) Low-threshold mechanical threshold: von Frey test (Control, n = 8; NaV1.6Nav1.8 KO, n = 10). (c) High-threshold mechanical threshold: Randall Sellito test (Control, n = 5; NaV1.6Nav1.8 KO, n = 9). (d) Noxious heat (spinal reflex) threshold: Hargreaves test apparatus (Control, n = 8; NaV1.6Nav1.8 KO, n = 10). (e) Noxious heat thresholds (Supraspinal response) measured using hot plate test at 52 °C and 56 °C (Control, n = 12; NaV1.6Nav1.8 KO, n = 16). (f) Dynamic cold plate test: the number of jumps/paw lifts was measured as temperature of the plate is changed from 25 °C to 0.5 °C (Control, n = 4; NaV1.6Nav1.8 KO, n = 9). (j) Thermal place preference test: the percentage of time spent at a test temperature (20 °C, 28 °C, 38 °C or 46 °C) versus control temperature 33 °C (Control, n = 7; NaV1.6Nav1.8 KO, n = 9). (g) Noxious cold thresholds: cold plate test at 0.5 °C (Control, n = 10; NaV1.6Nav1.8 KO, n = 10). (h) Cooling: dry ice test (Control, n = 5; NaV1.6Nav1.8 KO, n = 9). (i) Cooling: acetone test (Contorl, n = 5; NaV1.6Nav1.8 KO, n = 9). Color code: Control, white; NaV1.6Nav1.8 KO, orange. Data are presented as mean ± SEM. No difference (p > 0.05) was observed based on unpaired t test.
Previous reports suggested the involvement of NaV1.6 in chemotherapy induced acute cooling-aggravated pain23. Our data showed that NaV1.6Nav1.8 KO mice and heterozygous controls had similar withdrawal latency in all acute cold pain tests, including cold plate test (0.5 °C), dynamic cold plate test (temperature dropped from 25 °C to 0.5 °C at 5 °C/min), dry ice test and acetone test (Fig. 3g–j).
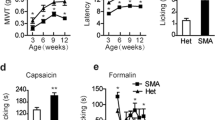
We then examined the impact of Nav1.8 Cre-driven NaV1.6 knockout on inflammatory and neuropathic pain. Intra-plantar injections of formalin or complete Freund’s adjuvant (CFA) were performed to induce acute or long-term hind paw inflammation. Behavioral responses in early and late phases of formalin test in NaV1.6Nav1.8 KO mice were similar to those in control mice (Fig. 4a,b). Both control and NaV1.6Nav1.8 KO mice displayed CFA-induced mechanical hypersensitivity, which lasted for more than 7 days before the withdrawal thresholds returned to baseline values (Fig. 4c). Similarly, the NaV1.6Nav1.8 KO mice developed CFA-induced thermal hypersensitivity as their control littermates (Fig. 4d).

Inflammatory and neuropathic pain behaviors in NaV1.8-Cre driven NaV1.6 KO mice. (a,b) Behavioral responses of Nav1.8-Cre driven Nav1.6 KO mice (NaV1.6Nav1.8 KO, n = 7) and heterozygous control littermates (Control, n = 7) following intra-plantar injection of 5% formalin (10 µl). (c) Mechanical sensitivity measured by von Frey test following intra-plantar CFA injection (Control, n = 7; NaV1.6Nav1.8 KO, n = 9). (d) Thermal sensitivity measured by Hargreaves test following intra-plantar CFA injection. (Control, n = 7; NaV1.6Nav1.8 KO, n = 9). (e) Mechanical sensitivity measured by von Frey test following SNI (Control, n = 8; NaV1.6Nav1.8 KO, n = 10). (f) Thermal sensitivity measured by Hargreaves test following SNI (Control, n = 8; NaV1.6Nav1.8 KO, n = 10). Color code: Control, black; NaV1.6Nav1.8 KO, orange. Data are presented as mean ± SEM. No difference (p > 0.05) was observed based on unpaired t test.
To study the impact of NaV1.6 knockout in NaV1.8-positive neurons on neuropathic pain behavior, we used the SNI model, in which the common peroneal and the sural branches of the sciatic nerve were transected and the tibial branch was left intact35. Following SNI, mechanical pain threshold was measured weekly using von Frey filaments. NaV1.6Nav1.8 KO mice developed mechanical allodynia to the same extent as their heterozygous control littermates as early as week one and maintained this lower threshold for four weeks post-surgery (Fig. 4e). As previously described using this model35, NaV1.6Nav1.8 KO mice did not show any abnormality in thermal thresholds (Fig. 4f).
AAV-mediated NaV1.6 KO attenuates SNI-induced mechanical allodynia
The normal pain behavior of the NaV1.6Nav1.8 KO mice might be the result of several factors including compensatory effects during development and the limited knockout of NaV1.6 to the NaV1.8-positive neuronal population. To further explore the contribution of NaV1.6 to neuropathic pain, we used an adeno associated virus (AAV)-mediated approach to achieve adult-onset knockout of the channel that was not limited to NaV1.8-positive neurons DRG neurons. The advantages of using this approach include (1) the deletion of NaV1.6 takes place in adult mice so that any developmental compensation could be minimized; (2) AAV targets both NaV1.8-positive and NaV1.8-negative neurons; and (3) the AAV strain (AAV2/5) used in this study was previously characterized to efficiently and selectively infect primary sensory neurons when delivered intrathecally36,37.
AAV2/5-Cre or control AAV2/5-GFP were injected intrathecally into NaV1.6flox/flox tdTomato mice. AAV2/5-Cre infection induces total NaV1.6 knockout in these neurons and produces red fluorescent protein to mark the affected neurons (NaV1.6AAV KO), whereas neurons infected with AAV2/5-GFP will retain homozygous WT NaV1.6 alleles and are marked with green fluorescence (WT Control). We have confirmed in this study that intrathecal administration of AAV2/5-Cre specifically targets DRG neurons and does not infect spinal dorsal horn neurons (Supplemental Fig. 3).
To assess the efficiency of viral infection in DRG neurons, we used GFP/NeuN or tomato/NeuN double labelling to determine the percentage of AAV2/5 infected cells and showed that intrathecal injection of AAV2/5 produced a high infection rate (50 to 80%) in L3 to L5 DRG neurons (Fig. 5a). Unlike NaV1.8-driven Cre expression which was observed predominantly in small neurons and limited to a small proportion of large neurons, AAV2/5-Cre was expressed in a substantial fraction of large neurons (Fig. 5b) (AAV-GFP: expressed in 245 out of 485 L4 DRG neurons, n = 3; AAV-Cre: expressed in 275 out of 502 L4 DRG neurons, n = 4).

AAV-Cre mediated NaV1.6 knockout in adult mice attenuates SNI-induced mechanical allodynia. (a) Representative images of L4 DRG sections showing AAV-GFP and AAV-Cre infected DRG neurons, which are marked by GFP and tdTomato, respectively. Neurons were identified by the neuronal marker NeuN. Scale bar = 50 µm. (b) Size distribution of AAV-GFP and AAV-Cre infected DRG neurons. (AAV-GFP: expressed in 245 out of 485 L4 DRG neurons, n = 3; AAV-Cre: expressed in 275 out of 502 L4 DRG neurons, n = 4). (c) Noxious heat thresholds measured by hot plate test in AAV-GFP control (WT control, n = 8) and AAV-Cre mediated NaV1.6 knockout (NaV1.6AAV KO, n = 15). (d) Noxious heat thresholds: Hargreaves test (WT control, n = 7; NaV1.6AAV KO, n = 11). (e) Noxious cold thresholds: cold plate test (WT control, n = 5; NaV1.6AAV KO, n = 7). (f) Noxious mechanical thresholds measured using von Frey test (WT control, n = 6; NaV1.6AAV KO, n = 11). (g) Mechanical allodynia following SNI was measured by the von Frey test in WT control (n = 10) and Nav1.6AAV KO mice (n = 12). Difference between the two groups was significant at week 1 (p = 0.0226), week 4 (p = 0.0455) and week 6 (p = 0.0336) post-surgery. (h) Thermal threshold measured by Hargreaves test was not changed compared to baseline in both WT control (n = 14) and Nav1.6AAV KO mice (n = 15). Data are presented as mean ± SEM. *p < 0.05 based on two-way ANOVA followed by Bonferroni post hoc test.
To determine the functional consequences of AAV-mediated knockout of NaV1.6 on acute pain thresholds, we assessed thermal and mechanical thresholds using hot/cold plate test, Hargreaves test and von Frey test, 3 weeks following viral vector injection, a time delay known to result in peak viral infection36. Compared to control animals injected with AAV2/5-GFP, NaV1.6AAV KO mice exhibited similar behavior in all these three tests (Fig. 5c–f). We then investigated the effect of AAV-Cre mediated NaV1.6 knockout on neuropathic pain behavior. Intrathecal injection of AAV was conducted at the same time as SNI surgery. Thermal and mechanical thresholds were assessed after surgery once per week for six weeks using Hargreaves test and von Frey test, respectively. Compared with animals receiving control AAV2/5-GFP, NaV1.6AAV KO mice showed a partial reduction in SNI-induced mechanical allodynia. The attenuation of mechanical allodynia was statistically significant at week 1 (p = 0.0226, two-way ANOVA followed by Bonferroni post hoc test), week 4 (p = 0.0455) and week 6 (p = 0.0336) post-surgery (Fig. 5g). As previously shown, thermal sensitivity was unaffected in this neuropathic pain model (Fig. 5h).
AAV-mediated NaV1.6 knockout interrupts NaV1.6 accumulations at the site of injury
To investigate the molecular and cellular basis of how AAV-Cre mediated NaV1.6 deletion attenuates injury-induced mechanical allodynia, we studied local NaV1.6 distribution at the site of injury 6 weeks after SNI. In intact nerves, NaV1.6 staining was seen along C-fibers and at nodes of Ranvier in A-fibers (Fig. 6a.I). Following SNI, NaV1.6 staining was reduced in C-fibers but the presence of NaV1.6 was retained at all nodes in the neuroma (Fig. 6a.IV). Importantly, the density of nodes was significantly higher in the neuroma (both common peroneal and sural nerves) than in intact tibial nerve (intact: 792/mm2; neuroma: 3003/mm2; n = 10; p < 0.001, unpaired t test) (Fig. 6b). The average length and width of the nodal gap in neuroma were significantly different from those in intact nerve (length × width in intact nerve: 0.77 ± 0.03 µm × 1.58 ± 0.06 µm; neuroma: 0.94 ± 0.03 µm × 0.88 ± 0.03 µm; n = 10; p < 0.001, unpaired t test) (Fig. 6c,d). These anatomical changes may represent newly formed nodes on regenerating fibers within the neuroma.

NaV1.6 knockout leads to empty nodes in myelinated fibers within the neuroma. (a) Confocal images showing NaV1.6 or panNaV (red) and contactin-associated protein (caspr, green) immunostaining as well as tdTomato signal (blue) in intact Tibial nerve (upper row) or neuroma (Common Peroneal nerve, bottom row) from AAV-GFP infected (WT control) and AAV-Cre infected (NaV1.6AAV KO) mice. Samples were collected 6 weeks after SNI and AAV injection. In intact nerve (aI) and neuroma (aIV) from wild-type mice, NaV1.6 protein is present in every node of Ranvier (filled arrowhead). In intact nerve from NaV1.6AAV KO mice NaV1.6 (aII) protein is stable in nodes (unfilled arrow head) along tdTomato positive fibers (blue, reporter for NaV1.6 gene excision); similar nodal staining along tdTomato positive fibers is observed when a pan sodium channel antibody is used (aIII, unfilled arrow head). In neuroma from NaV1.6AAV KO mice (aV), NaV1.6 protein is only present at nodes (filled arrow head) along tdTomato-negative fibers but absent at nodes along tdTomato-positive (blue) fibers (unfilled arrow head). Using a pan sodium channel antibody shows that the absence of Nav1.6 protein at nodes (unfilled arrow head) along tdTomato-positive (blue) fibers is not compensated by other NaV isoforms (aVI). Filled arrow heads point to representative nodes along tdTomato-negative A-fibers and unfilled arrow heads point to representative nodes along tdTomato-positive A-fibers. Arrows point to representative non-myelinated regions within the neuroma where sodium channels accumulate following injury. Scale bar = 10 µm; Inset scale bar = 2 µm. (b) Comparison of average density of NaV1.6-positive nodes in intact nerve (white, n = 10) and neuroma (grey, n = 10). (c) Comparison of average nodal gap length in intact nerve (white, n = 10) and neuroma (grey, n = 10). (d) Comparison of average nodal gap width in intact nerve (white, n = 10) and neuroma (grey, n = 10). (e) Percentage of NaV1.6-positive neurons in neuroma after AAV-Cre (NaV1.6AAV KO, n = 5) treatment or AAV-GFP (WT control, n = 5) treatment. Data are presented as mean ± SEM. ***p < 0.001 based on unpaired t test.
In NaV1.6AAV KO mice, nodal NaV1.6 staining was absent in tdTomato-positive fibers within neuroma (Fig. 6a.V). The percentage of nodes at neuroma which retained NaV1.6 was significantly reduced to 26.0 ± 2.0% in NaV1.6AAV KO mice compared to wild-type controls (n = 5, p < 0.001, upaired t-test) (Fig. 6e). Interestingly, NaV1.6 signal at nodes in intact tibial nerves was not diminished for up to 6 weeks when NaV1.6 gene was disrupted by AAV2/5-Cre expression (Fig. 6a.II and III), suggesting that, once inserted at nodes, NaV1.6 proteins are highly stable, presumably because of the formation of a nodal complex with long turn-over cycle. Using a pan sodium channel antibody, we did not observe any nodal staining along tdTomato-positive fiber (Fig. 6a.VI), indicating that other sodium channel isoforms were not present at NaV1.6-negative nodes within neuroma. However, pan Nav staining was observed along non-myelinated axons at the neuroma (Fig. 6a.VI, arrows), which was not seen using the NaV1.6 specific antibody (Fig. 6a.V), suggesting the accumulation of other sodium channel isoforms in these unmyelinated fibers38.
NaV1.6 knockout attenuates DRG neuron excitability following SNI
It has been previously established that both axotomized and intact neurons can become hyper-excitable following nerve injury39. We examined here whether knockout of NaV1.6 reduces excitability of DRG neurons. Current-clamp recordings were performed on both small and large DRG neurons cultured from NaV1.6AAV KO (marked by red fluorescence) or control (WT, marked by green fluorescence) mice that received SNI surgery.
Representative action potential traces from a large AAV-GFP infected WT DRG neuron (WT) in response to graded membrane potential depolarizations showed that an overshooting action potential was produced in this cell at a threshold of 950 pA (Fig. 7a). Representative action potentials recorded from a large NaV1.6AAV KO DRG neuron are shown in Fig. 7b. An action potential did not occur in this cell until the current stimulus reached a threshold of 2445 pA. The current threshold for action potential firing differed significantly in large WT versus NaV1.6AAV KO DRG neurons (Fig. 7c, Table 1), but were comparable in small DRG neurons (Fig. 7d, Table 2). The absence of NaV1.6 channels doubled the current threshold as compared to WT (WT, 918 ± 262 pA, n = 8; NaV1.6AAV KO, 2022 ± 252 pA, n = 13; p = 0.00941) in large DRG neurons (Fig. 7c, Table 1). Repetitive firing was suppressed in large NaV1.6AAV KO DRG neurons (Fig. 7e). In contrast, the maximal number of evoked action potentials in small DRG neurons was not different between control and NaV1.6AAV KO mice (Fig. 7f). There were no significant differences (p > 0.05) in resting membrane potential, input resistance, or action potential amplitude for small or large DRG neurons between wild-type control and NaV1.6AAV KO groups (Tables 1 and 2).

Absence of NaV1.6 decreases excitability of large DRG neurons following SNI. (a) Representative traces from a large DRG neuron expressing AAV-GFP (WT control), showing subthreshold responses to 900–945 pA current injections and subsequent all-or-none action potential by injection of 950 pA (current threshold for this neuron). (b) The same threshold protocol was applied to a large DRG neuron expressing AAV-CRE (NaV1.6AAV KO). The current threshold was 2445 pA for this neuron. Arrows with numbers indicate the current amplitude used to elicit the labeled response. (c) Comparison of current threshold in large WT control (n = 8) and NaV1.6AAV KO (n = 13) DRG neurons. (d) Comparison of current threshold in small WT control (n = 7) and NaV1.6AAV KO (n = 7) DRG neurons. (e) Scatter plot of maximal action potentials in response to external current stimuli up to 2000 pA in large WT control (n = 7) and NaV1.6AAV KO (n = 13) DRG neuron. (f) Scatter plot of maximal action potentials in response to external current stimuli up to 500 pA in small WT control (n = 6) and NaV1.6AAV KO (n = 7) DRG neurons. Data are presented as mean ± SEM. *p < 0.05 based on unpaired t test.
Discussion
Dynamic changes in NaV channel expression and function contribute to neuronal hyper-excitability following peripheral nerve injury40,41. Although NaV1.6 has recently been implicated in pain in humans27, and in animal models23,24,25,26, the specific contribution of NaV1.6 to the TTX-S current in DRG neurons and to neuropathic pain in knockout models has thus far not been reported. We report in this study that NaV1.6 contributes 34% of the TTX-S sodium current in small DRG neurons, 53% of this current in large NaV1.8-positive DRG neurons and 60% of this current in NaV1.8-negative DRG neurons. We report that NaV1.8-Cre driven knockout of NaV1.6 does not alter acute, inflammatory or neuropathic pain behaviors, while the adult-onset knockout of NaV1.6 via AAV-Cre ameliorates mechanical allodynia in the SNI model but without altering acute pain thresholds. We also show that SNI leads to an increase in the density of nodes of Ranvier in myelinated axons within the neuroma, and that NaV1.6 channels are stable at nodes for several weeks after the gene is knocked out. In parallel with the recovery of pain thresholds in the AAV-Cre mediated NaV1.6 knockout mice following SNI, excitability of large DRG neurons, but not small DRG neurons, was attenuated. Our data support an important role of NaV1.6 in large DRG neurons in neuropathic pain.
We have used three independent criteria to conclude that the expression of Cre recombinase induces the deletion of NaV1.6 floxed alleles in DRG neurons: (a) production of red fluorescence by a Cre-dependent tdtomato reporter cassette; (b) reduction of the TTX-S current and no change in the TTX-R current in neurons in which NaV1.6 is expected to be knocked out (red fluorescent neurons); (c) demonstration of the loss of nodal staining in myelinated fibers in which NaV1.6 is expected to be knocked out (red fluorescent fibers). The specificity of the NaV1.6 antibody was unequivocally validated in the mouse with global NaV1.6 knockout (Scn8amedtg, Supplemental Fig. 1). Taken together, this data supports our conclusion that the floxed NaV1.6 alleles are deleted in the DRG neurons that express Cre recombinase either from the NaV1.8 locus or via the AAV-Cre administration.
NaV1.6 is an abundant sodium channel in medium-large DRG neurons which produce myelinated A-fibers, and is also expressed in small DRG neurons that produce unmyelinated C-fibers42,43,44. We show here that deleting NaV1.6 in NaV1.8-positive neurons which represents 92% of small DRG neurons33 reduces the TTX-S current in these neurons by 34%. On the other hand, NaV1.6 channels contribute 47% of the TTX-S current in large NaV1.8-positive neurons and 60% in large NaV1.8-negative neurons. This functional data is consistent with molecular data using in situ hybridization which shows higher expression levels of NaV1.6 in medium and large DRG neurons over small DRG neurons43,45. Our functional data are also consistent with single-cell RNAseq profiling of mouse DRG neurons showing relatively higher NaV1.6 RNA in low threshold mechanoreceptors (LTMR) and proprioceptors, peptidergic nociceptor type 2 and C-LTMR, and very low levels in nonpeptidergic nociceptors46. The contribution of NaV1.6 to 34% of the TTX-S current in small NaV1.8-positive DRG neurons is consistent with the loss of 70% of the TTX-S current in small DRG neurons from NaV1.8-Cre driven knockout of NaV1.747. Thus, NaV1.6 and NaV1.7 appear to account for all the TTX-S current in small DRG neurons. In large DRG neurons, NaV1.6 channels contribute the majority of the TTX-S current, making up 53% of the TTX-S current in large NaV1.8-positive neurons and 60% in large NaV1.8-negative neurons. The balance of the TTX-S current in large DRG neurons after knocking out NaV1.6 is likely mediated by NaV1.1 and NaV1.71. The substantial contribution of NaV1.6 to TTX-S current in both small and large DRG neurons suggests a role for this channel in the transmission of pain signals.
The NaV1.8-Cre-driven NaV1.6 knockout mice were phenotypically indistinguishable from their littermate controls in acute, inflammatory and neuropathic pain models. The unaltered pain phenotype is unexpected given the reported role of NaV1.8-positive neurons in acute and inflammatory pain5,6,9,48, and the contribution of NaV1.6 to the amplitude of the compound action potential in the sciatic nerve44. Our observation suggests that in the absence of NaV1.6 in NaV1.8-positive neurons, other NaV isoforms are sufficient to mediate neuronal responses to noxious stimuli.
There are several alternative explanations for the unaltered pain thresholds when NaV1.6 deletion is limited to NaV1.8-positive neurons. Compensatory dysregulation of other molecules may have occurred during development, for example, the TTX-S current was up-regulated in NaV1.8-null mutant mice6, and NaV1.7 knockout was reported to increase met-enkephalin expression49. Additionally, NaV1.8 expression has been reported in CNS nuclei of naïve mice50, thus NaV1.8-Cre driven NaV1.6 knockout in these neurons may confound the effects of NaV1.6 deletion in primary afferents. Moreover, normal neuropathic pain has also been reported to be retained following NaV1.8 knockout, NaV1.7/NaV1.8 double knockout or genetic ablation of NaV1.8-positive neurons5,6,51. It is possible that NaV1.8-positive neurons, most of which are small DRG neurons, might have limited effect on neuropathic pain behavior.
AAV-mediated NaV1.6 knockout has adult onset and is not limited to NaV1.8-positive neurons, thus providing an opportunity to avoid the confounds of the NaV1.8-Cre approach to investigate the role of NaV1.6 in pain. Injured myelinated Aβ afferents generate most of the ectopic discharges around the time of pain onset52,53, and sodium current block in large myelinated fibers has been shown to suppress mechanical allodynia in a few neuropathic pain models54. We report here that AAV-mediated NaV1.6 knockout, likely in large neurons, significantly attenuates SNI-induced mechanical allodynia. The partial attenuation of pain hypersensitivity observed in this study should be considered an underestimation of the contribution of NaV1.6 to mechanical allodynia due to the incomplete knockout of NaV1.6 using this approach (range of infection rate: 50–80% of DRG neurons).
Traumatic injury to peripheral nerves can lead to the formation of painful neuromas, tangled masses of blind-ending axons and proliferating Schwann cells with connective tissue which can produce spontaneous pain, brush-evoked allodynia and pinprick hyperalgesia55. Ectopic firing arising from myelinated Aβ fibers within neuromas has been implicated in neuropathic pain56. The contribution of TTX-S channels, including NaV1.6, to ectopic firing at neuromas is supported by the findings that treatment with TTX at concentrations not likely to affect TTX-R channels reduces this ectopic activity57. We have shown here that NaV1.6 accumulates at nodes of Ranvier that form de novo in neuromas. A similar accumulation of NaV1.6 has been observed after infraorbital nerve lesion in a rat trigeminal pain model58. Although we did not determine the reduction in RNA levels of NaV1.6 in AAV-Cre treated mice, the functional loss of NaV1.6 at de novo nodes along tdTomato-positive fibers in the neuroma and increased threshold for action potential firing and reduced evoked firing in large DRG neurons following SNI injury is consistent with the knockout of the floxed NaV1.6 alleles in these mice. Our data also suggest that NaV1.6 is the only isoform expressed at the newly formed nodes within neuromas, because we did not detect a signal at these nodes using a pan-sodium channel antibody. NaV1.6 immunostaining remained at nodes in intact Tibial fibers for up to 6 weeks following NaV1.6 deletion, the longest time tested in this study, reflecting the stability of NaV1.6 channels once inserted at nodes. Taken together, these data suggest that new nodal formation at neuroma and NaV1.6 accumulation at these nodes likely contribute to the development of injury-induced neuropathic pain.
Injury causes an increase in the excitatory drive which is manifested as hyperexcitability of DRG neurons that underlie pain. We show in this study an increased density of nodes of Ranvier at the neuroma with a robust nodal NaV1.6 signal, comparable to that observed at nodes of uninjured fibers, and short intermodal distance. We also observed the absence of KV1.1/KV1.2 at juxta-paranodes (Supplemental Fig. 4). The significant reduction of KV1.1/KV1.2 in myelinated fibers in the mouse SNI model is consistent with a recent report of dynamic redistribution of KV1 channels within nodes following injury59. The increased nodal density with normal NaV1.6 levels, and the loss of KV1.1/KV1.2 at the juxta-paranodes are predicted to increase the safety factor for reliability and temporal fidelity of action potential conduction at the neuroma60. These changes enhance the excitatory drive which is predicted to contribute to mechanical allodynia in the SNI model (Fig. 8).

Schematic illustration of myelinated axons in intact nerve and regenerating myelinated axons within the neuroma. In intact sciatic nerve (the Tibial nerve in the SNI model), NaV1.6 is clustered at nodes of Ranvier with KV1.1 and KV1.2 present at juxta-paranodal regions. Regenerating myelinated axons within the neuroma show higher density of nodes of Ranvier with NaV1.6 as the only NaV isoform present at these nodes, and are characterized by elongated nodal length and smaller fiber caliber and loss of KV1.1 or KV1.2 at juxta-paranodal regions. The increased nodal density with the accumulation of NaV1.6 at these nodes and the loss of KV1.1 and KV1.2 at the juxta-paranodes, and the shorter intermodal distance increase the probability of secure conduction of action potentials that are initiated in the injured fibers and the probability of burst and spontaneous firing, contributing to hyperexcitability of DRG neurons and mechanical allodynia.
We have previously shown that NaV1.3, NaV1.7 and NaV1.8 channels accumulate within blind-ends of injured axons in painful human neuromas and in a rat model and suggested that they contribute to ectopic at the neuroma38,61. We provide here clear evidence for markedly increased density of nodes of Ranvier, reducing the intermodal distance, with dense nodal staining for NaV1.6 at the site of nerve injury. Reduced intermodal distances are known to increase conduction safety factor and can contribute to repetitive firing62. We suggest that the ectopic action potential initiation that may arise at the nerve endings supported by the accumulation of sodium channels, has a higher probability of propagating along the injured myelinated fibers because of the closed spacing and accumulation of NaV1.6 at nodes within the neuroma, and thus contribute to afferent hyperexcitability in the injured nerve.
In conclusion, we report here that NaV1.6 contributes substantially to the TTX-S current in large and small DRG neurons. NaV1.6 knockout in NaV1.8-positive neurons does not alter acute, inflammatory or neuropathic pain behaviors. By contrast, AAV-mediated NaV1.6 knockout in adult mice partially attenuates SNI-induced mechanical allodynia as early as one week following injury. In parallel with ameliorated mechanical pain, we show that AAV-Cre mediated NaV1.6 knockout prevents NaV1.6 accumulation at de novo formed nodes of Ranvier adjacent to the site of nerve injury, and reduces excitability of large DRG neurons. Taken together, these data provide strong evidence for a role of NaV1.6 in large neurons in neuropathic pain.
Materials and Methods
Animals and surgical procedures
All animal experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the IACUC of the Veterans Administration Connecticut Healthcare System. Adult mice (2 to 4 months) of both sexes were used in this study. Mouse strains with floxed Scn8a (NaV1.6flox/flox)32 and Scn8amedtg (global NaV1.6 KO)28 were a generous gift from Dr. Miriam Meisler (University of Michigan). NaV1.6 global knockout mice survive up to two weeks, and the homozygous KO mice become visually identifiable around P12 (smaller size and hindlimb paralysis), whereas wildtype and heterozygous mice are phenotypically indistinguishable. Scn8amedtg mice and their wildtype littermates were genotyped before harvesting their tissues. NaV1.8-Cre driven NaV1.6 knockout (NaV1.8+/Cre, NaV1.6flox/flox) and heterozygous (NaV1.8+/Cre, NaV1.6flox/+) mice (21 to 33 g) were used in one set of experiments. In the other set of experiments, homozygous NaV1.6-floxed (NaV1.6flox/flox) mice (21 to 33 g) were separated into two groups which received intrathecal injection of AAV viruses that carry the Cre recombinase (AAV-Cre) or GFP (AAV-GFP) to generate AAV-Cre mediated NaV1.6 knockout and wild-type control mice. All mice carried a Cre-reporter cassette (loxP-stop-loxP-tdTomato fluorescent protein), which leads to the production of a red fluorescent protein in neurons that express a functional Cre recombinase33.
Intrathecal injection
Under isoflurane (1–3% in oxygen) anesthesia, a laminectomy was performed between lumbar levels L5 and L6 to expose the thecal sac. This allowed visual confirmation that the needle entered the intrathecal space without damaging the spinal cord. To avoid injury to the underlying neural tissue, the needle was kept at midline and slowly inserted at a shallow angle (less than 30°) underneath the dura. Virus particles in a 5 μl volume were injected using a 10 μl Hamilton syringe with a 33-gauge needle. After injection, the paraspinal muscle and skin layers were closed using 6–0 monofilament nylon sutures (ETHICON). The viruses used in this study were as follows: AAV2/5-CMV-Cre, AAV2/5-CMV-eGFP (Gene Transfer Vector Core, IA). Virus titers were on the order of 1 × 1013 viral genomes (vg)/ml.
SNI
Procedures were performed as previously described35 immediately after the intrathecal injection of AAV. Briefly, an incision (1 cm) was made on the disinfected surgical field (left lateral mid-thigh) and the underlying muscles were separated to expose the three branches of the sciatic nerve. The common peroneal and sural branches were tightly ligated with 6-0 silk sutures (DemeTECH). The ligated branches were then transected and approximately 2 mm of the distal nerve stump was excised to minimize regeneration. Efforts were made to minimize any contact with the spared tibial branch. In sham animals, the sciatic nerve and its branches were exposed identically but were neither ligated nor transected. The muscle and skin layers were then closed with 6-0 monofilament nylon sutures (ETHICON).
Behavioral assays
All animals were acclimatized to the testing apparatus in three one-hour habituation sessions. One experimenter blind to genotypes or treatment groups performed all behavioral tests.
Rotarod test
To assess sensorimotor coordination, animals were placed onto the Rotarod apparatus running at 4 r.p.m., which was then accelerated to a maximum of 40 r.p.m. in 150 s. The time spent on the rotating rod was recorded with the cutoff time of 180 s.
von Frey test
To measure mechanical thresholds, mice were placed on an elevated wire grid and the plantar surface of the left paw (ipsilateral to the surgery/injection) of each animal was presented with a series of calibrated von Frey hairs (Stoeling, Wood Dale, IL). The 50% withdrawal threshold was determined using the “up-down” method63.
Randall-Sellito test
Noxious mechanical thresholds were measured using the Randall-Sellito electronic algesimeter (IITC 2500 Digital Paw Pressure Meter, IITC Life Science, Woodland Hills, CA). Each animal received 5 min of handling before being immobilized in the holding chamber. A uniformly increasing pressure was applied to the tail via a blunt conical probe until a withdrawal response resulted. The mechanical stimulation was repeated 3 times with 15 min separation between consecutive tests. The cut-off value was set at 500 g to prevent tissue damage.
Hargreaves test
To assess noxious heat threshold, the plantar paw surface was exposed to radiant heat using a Hargreaves apparatus (IITC, Woodland Hills, CA) and the paw withdrawal latency was measured. The heat stimulation was repeated 5 times with 15 min separation between consecutive tests. The cut-off value was set at 30 seconds to prevent tissue damage.
Hot/Cold plate
To assess thermal sensitivity, animals were placed on a metal plate with a constant temperature of 52 °C, 56 °C or 0.5 °C. The animal was removed from the plate immediately upon licking or flicking a hindpaw or if no response occurred within a predetermined cut-off time (30 s for hot plate and 120 s for cold plate).
Dynamic cold plate
To assess the noxious cold tolerance, animals were placed on the test arena with the surface temperature progressively cooled from 25 °C to 0.5 °C at a rate of −5 °C/min and their nocifensive behaviors (jumping and paw lifting) were noted.
Acetone test
50 µl of acetone was sprayed onto the plantar surface of the hindpaw using a blunt rubber tube attached to a 1-ml syringe. Behavioral responses were recorded over 30 s for each of five acetone applications. The responses were graded to a three-point scale: 0 = a brisk lifting, licking, or flicking of the hindpaw, which subsides immediately; 1 = lifting, licking, and/or flicking of the hindpaw, which continues beyond the initial application, but subsides within 5 s; 2 = protracted, repeated lifting, licking, and/or flicking of the hindpaw64.
Dry ice test
A dry ice pellet was made by compressing fine dry ice powder into a 3-ml syringe (with top cut off) and was then firmly pressed against the glass surface underneath the mouse hind paw until a withdrawal response resulted65. The testing procedure was repeated 3 times with 15 min separation between consecutive tests.
Two-plate thermal preference test
The thermal preference apparatus consisted of two cold/hot plates (Bioseb, Chaville, France) placed side-by-side and enclosed in a plexiglass chamber. The reference plate was set to 33 °C, whereas the test plate was adjusted to the specific temperature of 20 °C, 28 °C, 38 °C or 46 °C. Animals were gently placed at the center of the apparatus and could move freely between plates. Movements were recorded for 600 s by an infrared camera mounted directly above the enclosed space. The percentage of the time spent on the test plate was recorded with an automated video tracking system.
Formalin test
For induction of short-term inflammation, 10 µl of 5% formalin solution was injected subcutaneously into the left hind paw (intraplantar). The spontaneous nocifensive responses (licking, flicking, lifting the injected paw) were recorded for 50 min post injection.
CFA injections
Mice were anaesthetized by exposure to isoflurane (1–3%), administered by a calibrated vaporizer. For induction of long-term inflammation, 10 µl of complete Freund’s adjuvant (CFA; Sigma, containing 1 mg/ml of Mycobacteriom tuberculosis) were injected into the right hind paw using a 10 µl-Hamilton syringe (Hamilton company, Reno, Nevada).
Immunohistochemistry
Mice were anesthetized with intraperitoneal ketamine/xylazine injection (100/10 mg/kg) and transcardially perfused with 0.01 M PBS (pH 7.4) followed by ice-cold 4% paraformaldehyde in 0.14 M Sorensen’s phosphate buffer (pH 7.4). Tissues (sciatic nerve, L3, L4 and L5 DRG and spinal cord) were removed, immersion-fixed in 4% paraformaldehyde (total fixation time 20 min) and cryo-protected with 30% (w/v) sucrose in PBS overnight at 4 °C. Tissue sections were cut on a cryostat at 6 µm (sciatic nerve), 10 µm (DRG), or 20 µm (Spinal Cord) and mounted on slides (Fisher Scientific, Pittsburgh, PA). Sections were immediately processed for detection of target protein or stored at −20 °C for future use.
Sections were incubated in the following solutions: (1) blocking solution (PBS containing 4% normal donkey serum, 2% BSA, 0.1% Triton X-100, and 0.02% sodium azide) for 1 h at room temperature; (2) primary antibodies guinea pig anti-contactin-associated protein (caspr)66 (1:2000); rabbit anti-NaV1.6 (1:250, Millipore); rabbit anti-panNaV (1:250, Millipore); chicken anti-GFP (1:500, Abcam); mouse anti-NeuN Alex Fluor 488 conjugated (1:500, Millipore); in blocking solution at 4 °C overnight; (3) PBS, 3 × 10 min each; (4) secondary antibodies from Jackson Immuno Research Lab at 1:500 dilution in blocking solution for 1 h at room temperature; (5) PBS, 3 × 10 min. Tissue sections were examined with a Nikon Eclipse E800 fluorescence microscope or a Nikon C1 confocal microscope (Nikon USA, Melville, NY). The specificity of NaV1.6 staining described in this study was validated in Scn8amedtg mice (global NaV1.6 KO) (Supplementary Fig. 2).
Isolation of DRG neurons
DRG neurons were isolated using protocols described by Dib-Hajj et al. 2009 and Huang et al. 201367,68. Briefly, DRG tissue was first incubated at 37 °C for 20 min in complete saline solution (CSS) [in mM: 137 NaCl, 5.3 KCl, 1 MgCl2, 25 sorbitol, 3 CaCl2, and 10 Hepes, pH 7.2 adjusted with NaOH] supplemented with 0.5 U/ml Liberase TM (Roche) and 0.6 mM EDTA. The tissue was then incubated for 15 min at 37 °C in CSS containing 0.5 U/ml Liberase TL (Roche), 0.6 mM EDTA, and 30 U/ml papain (Worthington Biochemical). DRGs were centrifuged and triturated in 0.5 ml of DRG culture medium containing 1.5 mg/ml BSA (low endotoxin) and 1.5 mg/ml trypsin inhibitor (Sigma). The cell suspension was filtered through 70 μm nylon mesh cell strainer (Becton Dickinson) to remove debris, and the mesh was then washed twice with 2 ml of DRG culture medium. DRG neurons were pelleted by centrifugation and resuspended in 1 ml of DRG culture medium. Aliquots of 100 µl cell suspension were seeded onto poly-D-lysine/laminin-coated coverslips (BD), and incubated at 37 °C in a 95% air/5% CO2 (vol/vol) incubator. After 45 min of incubation, 1.4 ml of DRG culture medium was added into each well. For current-clamp recording, nerve growth factor (50 ng/ml) and glial cell line-derived neurotrophic factor (50 ng/ml) were added to DRG culture medium.
Whole-cell electrophysiology
The protocols for recording action potential were described previously68. Briefly, voltage-clamp recordings were conducted at room temperature (22 ± 1 °C) within 24 hrs after DRG neuron isolation using an EPC-10 amplifier and Patchmaster program (v 53; HEKA Elektronik, Lambrecht/ Pfalz,Germany). Electrodes were fabricated from 1.6 mm outer diameter borosilicate glass micropipettes (World Precision Instruments, Sarasota, FL) and fire-polished using a Sutter Instruments P-97 puller (Novato, CA). Electrodes had a resistance of 0.5–1.2 MΩ. Pipette potential was adjusted to zero before seal formation, and liquid junction potential was not corrected. We implemented 80–90% series resistance compensation to reduce voltage-errors. Linear leak currents were subtracted using the P/N method69. We allowed 5 min equilibration period once whole-cell configuration was achieved before we started recording sodium currents. We acquired data at 50 kHz, and filtered it with a low-pass Bessel setting of 2.9 kHz.
We first measured the TTX-S sodium currents in NaV1.8-Cre driven NaV1.6 knockout (NaV1.8+/Cre, NaV1.6flox/flox) and heterozygous (NaV1.8+/Cre, NaV1.6flox/+) mice using whole-cell voltage-clamp technique. The pipette solution for voltage-clamp recordings in small neurons contained (in mM): 140 CsF, 10 NaCl, 1 EGTA, 10 dextrose and 10 HEPES, pH 7.3 with CsOH (308 mOsmol/L adjusted with sucrose). The extracellular bath solution for voltage-clamp recording contained (in mM): 20 NaCl, 120 choline chloride, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 5 CsCl, 20 Tetraethylammonium chloride (TEA·Cl), pH 7.32 with NaOH (327 mOsmol/ L). The bath solution contained 0.1 mM CdCl2 to block calcium currents and 1 mM 4-Aminopyridine to block potassium currents. Small DRG neurons were held at −100 mV. Total voltage-gated sodium current was measured with 100 ms step depolarizations (−80 to +30 mV) in 5 mV increments (5 s intervals) from the holding potential. The pipette solution for voltage-clamp recordings in large neurons contained (in mM): 140 CsCl, 10 NaCl, 0.5 EGTA, 3 Mg-ATP, and 5 Hepes, pH 7.31 with CsOH (310 mOsmol/L adjusted with dextrose). DRG neuronal membrane is usually more stable with CsF-based pipette solutions which is why CsF is commonly used for routine recordings and for long recording protocols. However, sodium currents are much bigger in large diameter than small diameter neurons and the use of CsCl permits a better clamping of large currents. The extracellular bath solution contained (in mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 Hepes, 5 CsCl, 20 tetraethylammonium chloride (TEA·Cl), 0.1 CdCl2, 5 4-aminopyridine, pH 7.30 with HCl (320 mOsmol/L adjusted with dextrose). Large DRG neurons were held at −70 mV. Current-voltage (I-V) relationship was determined using 100-ms step depolarizations (−65 to +40 mV) in 5 mV increments (5 s intervals) from a 500 ms prepulsing potential at −100 mV.
We performed voltage-clamp studies in large neurons from global NaV1.6 null mice (SCN8amedtg) and their wild-type littermates. The pipette solution contained (in mM): 140 CsF, 10 NaCl, 1 EGTA, 10 dextrose and 10 HEPES, pH 7.3 with CsOH (308 mOsmol/L adjusted with sucrose). Because we recorded resurgent current in these neurons, we decided to use CsF instead for the benefit of maintaining stable cell membrane during lengthy recordings. The bath solution contained (in mM): 30 NaCl, 110 choline chloride, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 5 CsCl, 20 Tetraethylammonium chloride (TEA·Cl), pH 7.33 with NaOH (339 mOsmol/l). Cells were held at −100 mV followed by a series of 100-ms depolarizations from −70 to +10 mV in 5 mV increments. To determine resurgent sodium currents, we used a strong depolarization (20 ms at +10 mV) followed by 200 ms intermediate repolarization pulses to voltages ranging from −10 to −80 mV in 5 mV increments.
TTX (1 µM) was added to the bath solution to isolate the TTX-R currents in both small and large neurons. TTX-S sodium currents in each neuron were obtained by subtracting the TTX-R currents from the total sodium current. Current density was determined by normalizing peak transient currents (I Trans ) with cell capacitance.
The protocols for current-clamp recordings were previously described68. Briefly, electrodes had a resistance of 0.7–1.5 MΩ. The pipette solution contained (in mM): 140 KCl, 0.5 EGTA, 5 Hepes, and 3 Mg-ATP (pH 7.30 adjusted with KOH and 310 mOsmol/L adjusted with dextrose). The extracellular solution contained (in mM): 140 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 10 Hepes (pH 7.30 adjusted with NaOH and 320 mOsmol/L adjusted with dextrose). We established the whole-cell configuration in the voltage-clamp mode before switching to the current-clamp mode. We included in our analysis cells with stable RMP (<10% variation) negative than −35 mV and overshooting action potentials (>85 mV RMP-to-peak). Input resistance was determined by the slope of a linear fit to hyperpolarizing responses to current steps from −5 to −40 pA in 5 pA increments. Threshold was determined at the first action potential in response to depolarizing current injections (200 ms) in 5 pA increments. Firing frequency was determined by the number of action potentials elicited in response to depolarizing stimuli (500 ms) from 25 to 500 pA in 25 pA increments for small DRG neurons, and from 100 to 2000 pA in 100 pA increments for large DRG neurons. Endogenous NaV1.8 currents were assessed by holding neurons at −50 mV70. DRG neurons that expressed NaV1.8 currents <1 nA and did not generate all-or-none action potentials in response to 200 ms current stimulus were excluded from analysis71.
Statistical analysis
For behavioral studies, we used two-way repeated analysis of variance (ANOVA) followed by Bonferroni post hoc test to assess differences in mechanical and thermal sensitivity between two treatment groups. For immunohistochemical studies, unpaired t-test was used to determine the significance of changes (p < 0.05). For electrophysiological data analysis, we used Fitmaster (HEKA Elektronik) and Origin (Microcal Software, Northampton, MA). Unless otherwise noted, statistical significance was determined (p < 0.05) using an independent t-test. Mann-Whitney test was used in comparison of firing frequencies in response to external current injections. Results are presented as mean ± Standard error of means (SEM).
References
Dib-Hajj, S. D., Cummins, T. R., Black, J. A. & Waxman, S. G. Sodium channels in normal and pathological pain. Annu Rev Neurosci 33, 325–347, https://doi.org/10.1146/annurev-neuro-060909-153234 (2010).
Toledo-Aral, J. J. et al. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proceedings of the National Academy of Sciences of the United States of America 94, 1527–1532 (1997).
Akopian, A. N., Sivilotti, L. & Wood, J. N. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 379, 257–262, https://doi.org/10.1038/379257a0 (1996).
Dib-Hajj, S. D., Tyrrell, L., Black, J. A. & Waxman, S. G. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proceedings of the National Academy of Sciences of the United States of America 95, 8963–8968 (1998).
Abrahamsen, B. et al. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science (New York, N.Y.) 321, 702–705, https://doi.org/10.1126/science.1156916 (2008).
Akopian, A. N. et al. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nature neuroscience 2, 541–548, https://doi.org/10.1038/9195 (1999).
Amaya, F. et al. The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. The Journal of neuroscience: the official journal of the Society for Neuroscience 26, 12852–12860, https://doi.org/10.1523/jneurosci.4015-06.2006 (2006).
Priest, B. T. et al. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proceedings of the National Academy of Sciences of the United States of America 102, 9382–9387, https://doi.org/10.1073/pnas.0501549102 (2005).
Nassar, M. A. et al. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proceedings of the National Academy of Sciences of the United States of America 101, 12706–12711, https://doi.org/10.1073/pnas.0404915101 (2004).
Faber, C. G. et al. Gain of function Nanu1.7 mutations in idiopathic small fiber neuropathy. Annals of neurology 71, 26–39, https://doi.org/10.1002/ana.22485 (2012).
Faber, C. G. et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proceedings of the National Academy of Sciences of the United States of America 109, 19444–19449, https://doi.org/10.1073/pnas.1216080109 (2012).
Huang, J. et al. Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain 137, 1627–1642, https://doi.org/10.1093/brain/awu079 (2014).
Huang, J. et al. Sodium channel NaV1.9 mutations associated with insensitivity to pain dampen neuronal excitability. The Journal of Clinical Investigation 127, 2805–2814, https://doi.org/10.1172/JCI92373 (2017).
Han, C., Huang, J. & Waxman, S. G. Sodium channel Nav1.8: Emerging links to human disease. Neurology 86, 473–483, https://doi.org/10.1212/wnl.0000000000002333 (2016).
Dib-Hajj, S. D., Black, J. A. & Waxman, S. G. NaV1.9: a sodium channel linked to human pain. Nature reviews. Neuroscience 16, 511–519, https://doi.org/10.1038/nrn3977 (2015).
Dib-Hajj, S. D., Yang, Y., Black, J. A. & Waxman, S. G. The Na(V)1.7 sodium channel: from molecule to man. Nature reviews. Neuroscience 14, 49–62, https://doi.org/10.1038/nrn3404 (2013).
Bennett, D. L. & Woods, C. G. Painful and painless channelopathies. The Lancet. Neurology 13, 587–599, https://doi.org/10.1016/s1474-4422(14)70024-9 (2014).
Catterall, W. A., Kalume, F. & Oakley, J. C. NaV1.1 channels and epilepsy. The Journal of physiology 588, 1849–1859, https://doi.org/10.1113/jphysiol.2010.187484 (2010).
O’Brien, J. E. & Meisler, M. H. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Frontiers in genetics 4, 213, https://doi.org/10.3389/fgene.2013.00213 (2013).
Veeramah, K. R. et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. American journal of human genetics 90, 502–510, https://doi.org/10.1016/j.ajhg.2012.01.006 (2012).
Meisler, M. H. et al. SCN8A encephalopathy: Research progress and prospects. Epilepsia 57, 1027–1035, https://doi.org/10.1111/epi.13422 (2016).
Osteen, J. D. et al. Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 534, 494–499, https://doi.org/10.1038/nature17976 (2016).
Sittl, R. et al. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype NaV1.6-resurgent and persistent current. Proceedings of the National Academy of Sciences 109, 6704–6709, https://doi.org/10.1073/pnas.1118058109 (2012).
Deuis, J. R. a. b. et al. An animal model of oxaliplatin-induced cold allodynia reveals a crucial role for Nav1. 6 in peripheral pain pathways. Pain 154, 1749–1757 (2013).
Xie, W., Strong, J. A., Ye, L., Mao, J. X. & Zhang, J. M. Knockdown of sodium channel NaV1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. Pain 154, 1170–1180, https://doi.org/10.1016/j.pain.2013.02.027 (2013).
Xie, W., Strong, J. A. & Zhang, J. M. Local knockdown of the NaV1.6 sodium channel reduces pain behaviors, sensory neuron excitability, and sympathetic sprouting in rat models of neuropathic pain. Neuroscience 291, 317–330, https://doi.org/10.1016/j.neuroscience.2015.02.010 (2015).
Tanaka, B. S. et al. A Gain-of-Function Mutation in Nav1.6 in a Case of Trigeminal Neuralgia. Molecular Medicine 22, 338–348, https://doi.org/10.2119/molmed.2016.00131 (2016).
Burgess, D. L. et al. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nature genetics 10, 461–465, https://doi.org/10.1038/ng0895-461 (1995).
Blair, N. T. & Bean, B. P. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience 22, 10277–10290 (2002).
Vasylyev, D. V. & Waxman, S. G. Membrane properties and electrogenesis in the distal axons of small dorsal root ganglion neurons in vitro. J Neurophysiol 108, 729–740, https://doi.org/10.1152/jn.00091.2012 (2012).
Stirling, L. C. et al. Nociceptor-specific gene deletion using heterozygous NaV1.8-Cre recombinase mice. Pain 113, 27–36, https://doi.org/10.1016/j.pain.2004.08.015 (2005).
Levin, S. I. & Meisler, M. H. Floxed allele for conditional inactivation of the voltage-gated sodium channel Scn8a (NaV1.6). Genesis (New York, N.Y.: 2000) 39, 234–239, https://doi.org/10.1002/gene.20050 (2004).
Shields, S. D. et al. Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain 153, 2017–2030, https://doi.org/10.1016/j.pain.2012.04.022 (2012).
Cummins, T. R., Dib-Hajj, S. D., Herzog, R. I. & Waxman, S. G. Nav1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS letters 579, 2166–2170, https://doi.org/10.1016/j.febslet.2005.03.009 (2005).
Shields, S. D., Eckert Iii, W. A. & Basbaum, A. I. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. The Journal of Pain 4, 465–470, https://doi.org/10.1067/S1526-5900(03)00781-8 (2003).
Samad, O. A. et al. Virus-mediated shRNA knockdown of Na(v)1.3 in rat dorsal root ganglion attenuates nerve injury-induced neuropathic pain. Molecular therapy: the journal of the American Society of Gene Therapy 21, 49–56, https://doi.org/10.1038/mt.2012.169 (2013).
Tan, A. M., Samad, O. A., Dib-Hajj, S. D. & Waxman, S. G. Virus-Mediated Knockdown of Nav1.3 in Dorsal Root Ganglia of STZ-Induced Diabetic Rats Alleviates Tactile Allodynia. Molecular Medicine 21, 544–552, https://doi.org/10.2119/molmed.2015.00063 (2015).
Black, J. A., Nikolajsen, L., Kroner, K., Jensen, T. S. & Waxman, S. G. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Annals of neurology 64, 644–653, https://doi.org/10.1002/ana.21527 (2008).
Campbell, J. N. & Meyer, R. A. Mechanisms of neuropathic pain. Neuron 52, 77–92, https://doi.org/10.1016/j.neuron.2006.09.021 (2006).
Waxman, S. G. Acquired channelopathies in nerve injury and MS. Neurology 56, 1621–1627 (2001).
Waxman, S. G., Dib-Hajj, S., Cummins, T. R. & Black, J. A. Sodium channels and pain. Proceedings of the National Academy of Sciences of the United States of America 96, 7635–7639 (1999).
Ho, C. & O’Leary, M. E. Single-cell analysis of sodium channel expression in dorsal root ganglion neurons. Molecular and cellular neurosciences 46, 159–166, https://doi.org/10.1016/j.mcn.2010.08.017 (2011).
Black, J. A. et al. Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Brain research. Molecular brain research 43, 117–131 (1996).
Black, J. A., Renganathan, M. & Waxman, S. G. Sodium channel Na(v)1.6 is expressed along nonmyelinated axons and it contributes to conduction. Brain research. Molecular brain research 105, 19–28 (2002).
Fukuoka, T. et al. Comparative study of the distribution of the alpha-subunits of voltage-gated sodium channels in normal and axotomized rat dorsal root ganglion neurons. The Journal of comparative neurology 510, 188–206, https://doi.org/10.1002/cne.21786 (2008).
Usoskin, D. et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nature neuroscience 18, 145–153, https://doi.org/10.1038/nn.3881 (2015).
Vasylyev, D. V., Han, C., Zhao, P., Dib-Hajj, S. & Waxman, S. G. Dynamic-clamp analysis of wild-type human Nav1.7 and erythromelalgia mutant channel L858H. Journal of Neurophysiology 111, 1429–1443, https://doi.org/10.1152/jn.00763.2013 (2014).
Emery, E. C., Young, G. T., Berrocoso, E. M., Chen, L. & McNaughton, P. A. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science (New York, N.Y.) 333, 1462–1466, https://doi.org/10.1126/science.1206243 (2011).
Minett, M. S. et al. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nature communications 6, 8967, https://doi.org/10.1038/ncomms9967 (2015).
Lu, V. B., Ikeda, S. R. & Puhl, H. L. 3rd A 3.7 kb fragment of the mouse Scn10a gene promoter directs neural crest but not placodal lineage EGFP expression in a transgenic animal. The Journal of neuroscience: the official journal of the Society for Neuroscience 35, 8021–8034, https://doi.org/10.1523/jneurosci.0214-15.2015 (2015).
Nassar, M. A., Levato, A., Stirling, L. C. & Wood, J. N. Neuropathic pain develops normally in mice lacking both Na(v)1.7 and Na(v)1.8. Mol Pain 1, 24, https://doi.org/10.1186/1744-8069-1-24 (2005).
Boucher, T. J. et al. Potent analgesic effects of GDNF in neuropathic pain states. Science (New York, N.Y.) 290, 124–127 (2000).
Liu, C. N. et al. Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain 85, 503–521 (2000).
Xu, Z. Z. et al. Inhibition of mechanical allodynia in neuropathic pain by TLR5-mediated A-fiber blockade. Nature medicine 21, 1326–1331, https://doi.org/10.1038/nm.3978 (2015).
Nikolajsen, L., Black, J. A., Kroner, K., Jensen, T. S. & Waxman, S. G. Neuroma removal for neuropathic pain: efficacy and predictive value of lidocaine infusion. The Clinical journal of pain 26, 788–793 (2010).
Devor, M. Ectopic discharge in Abeta afferents as a source of neuropathic pain. Experimental brain research 196, 115–128, https://doi.org/10.1007/s00221-009-1724-6 (2009).
Omana-Zapata, I., Khabbaz, M. A., Hunter, J. C., Clarke, D. E. & Bley, K. R. Tetrodotoxin inhibits neuropathic ectopic activity in neuromas, dorsal root ganglia and dorsal horn neurons. Pain 72, 41–49 (1997).
Henry, M. A., Freking, A. R., Johnson, L. R. & Levinson, S. R. Sodium channel Nav1.6 accumulates at the site of infraorbital nerve injury. BMC neuroscience 8, 56, https://doi.org/10.1186/1471-2202-8-56 (2007).
Calvo, M. et al. Altered potassium channel distribution and composition in myelinated axons suppresses hyperexcitability following injury. eLife 5, e12661, https://doi.org/10.7554/eLife.12661 (2016).
Waxman, S. G. & Wood, S. L. Impulse conduction in inhomogeneous axons: effects of variation in voltage-sensitive ionic conductances on invasion of demyelinated axon segments and preterminal fibers. Brain research 294, 111–122 (1984).
Black, J. A. et al. Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J Neurophysiol 82, 2776–2785 (1999).
Waxman, S. G. & Brill, M. H. Conduction through demyelinated plaques in multiple sclerosis: computer simulations of facilitation by short internodes. Journal of neurology, neurosurgery, and psychiatry 41, 408–416 (1978).
Chaplan, S. R., Bach, F. W., Pogrel, J. W., Chung, J. M. & Yaksh, T. L. Quantitative assessment of tactile allodynia in the rat paw. Journal of neuroscience methods 53, 55–63 (1994).
Colburn, R. W. et al. Attenuated cold sensitivity in TRPM8 null mice. Neuron 54, 379–386, https://doi.org/10.1016/j.neuron.2007.04.017 (2007).
Brenner, D. S., Golden, J. P. & Gereau, R. W. A Novel Behavioral Assay for Measuring Cold Sensation in Mice. PloS one 7, https://doi.org/10.1371/journal.pone.0039765 (2012).
Rush, A. M. et al. Differential modulation of sodium channel Na(v)1.6 by two members of the fibroblast growth factor homologous factor 2 subfamily. The European journal of neuroscience 23, 2551–2562, https://doi.org/10.1111/j.1460-9568.2006.04789.x (2006).
Dib-Hajj, S. D. et al. Transfection of rat or mouse neurons by biolistics or electroporation. Nature protocols 4, 1118–1126, https://doi.org/10.1038/nprot.2009.90 (2009).
Huang, J. et al. Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience 33, 14087–14097, https://doi.org/10.1523/jneurosci.2710-13.2013 (2013).
Cummins, T. R., Rush, A. M., Estacion, M., Dib-Hajj, S. D. & Waxman, S. G. Voltage-clamp and current-clamp recordings from mammalian DRG neurons. Nature protocols 4, 1103–1112, https://doi.org/10.1038/nprot.2009.91 (2009).
Cummins, T. R. & Waxman, S. G. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. The Journal of neuroscience: the official journal of the Society for Neuroscience 17, 3503–3514 (1997).
Cheng, X. et al. Deletion mutation of sodium channel Na(V)1.7 in inherited erythromelalgia: enhanced slow inactivation modulates dorsal root ganglion neuron hyperexcitability. Brain 134, 1972–1986, https://doi.org/10.1093/brain/awr143 (2011).
Acknowledgements
We thank Dr. Edgardo Arroyo and Dr. Philip Effraim for helpful discussions, and Lawrence Macala and Palak Shah for technical support. Support for this research was provided by the Medical Research Service and Rehabilitation Research Service, Department of Veterans Affairs (SGW and SDH). The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University.
Author information
Authors and Affiliations
Contributions
L.C., J.H., S.G.W. and S.D.H. designed the experiments. L.C. and A.K.P. carried out in vivo experiments. J.H. and X.C. carried out electrophysiological experiments. L.C. carried out histological and imaging experiments. P.Z., F.D.H. and A.T. contributed to culture and tissue processing. L.C., J.H. and S.D.H. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, L., Huang, J., Zhao, P. et al. Conditional knockout of NaV1.6 in adult mice ameliorates neuropathic pain. Sci Rep 8, 3845 (2018). https://doi.org/10.1038/s41598-018-22216-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22216-w
This article is cited by
-
Piezo2 regulates colonic mechanical sensitivity in a sex specific manner in mice
Nature Communications (2023)
-
Neuroinflammation in the anterior cingulate cortex: the potential supraspinal mechanism underlying the mirror-image pain following motor fiber injury
Journal of Neuroinflammation (2022)
-
Concussion leads to widespread axonal sodium channel loss and disruption of the node of Ranvier
Acta Neuropathologica (2022)
-
Ion channel long non-coding RNAs in neuropathic pain
Pflügers Archiv - European Journal of Physiology (2022)
-
The physiological function of different voltage-gated sodium channels in pain
Nature Reviews Neuroscience (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
