
Abstract
Heading date (HD) and panicle length (PL) are important traits that affect rice breeding and are controlled by pleiotropic genes. Some alleles associated with HD and PL from wild relatives might differ from those in cultivated rice. In this study, a main effect HD quantitative trait locus from wild rice, qHD7.2, was identified using a chromosomal segment substitution line (CSSL) population. First, qHD7.2 was determined to be located near RM172 on chromosome 7 based on association analysis of phenotype data from six environments and 181 polymorphic molecular markers. CSSL39, which has the latest flowering of all CSSLs and carries qHD7.2, was selected for further study, and qHD7.2 was narrowed to a 101.1-kb interval using a CSSL39/9311 F2 population. An OsPRR37-homologous gene was found within this region. The wild type allele delayed flowering and shortened PL under long-day conditions. The HD7.2, which was identified as a candidate gene for qHD7.2, transcript level was substantially higher than that in 9311. Our data showed that HD7.2 is likely a novel OsPRR37 allele. Sequence analysis revealed that OsPRR37 in cultivated rice had multiple origins, and natural variation in the coding domain sequence and promoter region contribute to flowering time diversity in cultivated rice.
Similar content being viewed by others


Introduction
Asian cultivated rice varieties are distributed worldwide in a wide range of latitudes, from 53° north to 40° south; alternatively, its direct ancestor, common wild rice (Oryza rufipogon), is a short-day plant1. Artificial selection from naturally variable photoperiod sensitivity traits in rice germplasm is of paramount importance for rice global adaptation2. Heading date (HD) is a crucial determinant of rice diversification and domestication3,4. HD in rice is a typical quantitative trait locus (QTL) with complex inheritance, and is controlled by multiple genes. Clearly understanding the genetic basis of HD is important for elucidating the adaptation of rice to different cultivation areas and crop seasons.
Over the last two decades, hundreds of HD QTLs were documented in the Gramene database5 (http://archive.gramene.org/qtl/). Almost 65 HD genes have been cloned, and the photoperiod regulatory network are well documented. Hd3a (a rice orthologue of the Arabidopsis FT gene) and RFT1 (the closest homologue to Hd3a) are two florigen genes in the complex flowering time control network6,7, and the flowering-integrated factors Hd1 and Ehd1 receive signals from other genes to regulate flowering by affecting florigen gene expression8,9. Of these other genes, OsELF310, OsGI11, SE512, PhyB13, and Hd614 regulate the expression of Ghd715, Ehd316, OsLFL117, Hd16/EL118, RID119, and OsMADS5120 and belong to the Ehd1-dependent pathway. Additionally, several HD genes such as OsPRR3721, OsCO322, DTH223, and OsDof1224 directly control the expression of florigen genes independent of Hd1 and Ehd1. Molecular analysis revealed that natural variation in coding domain sequence and promoter sequences of some HD-QTL genes led to different responses to photoperiod sensitivity. Takahashi25 found that diverse combinations of natural variation in Hd1 proteins, Hd3a promoters, and Ehd1 expression levels contribute to flowering time diversity in the core collection of rice cultivars. Panicle length (PL) is an important trait which strongly affects yield components by determining panicle architecture in rice, such as grain number per panicle. PL can be used as a selection criterion for yield breeding and exhibits higher heritability than yield itself 26. Several QTLs associated with PL have been reported; among them, only dep1 and sp1 that lead to shorter panicles have been cloned27,28.
As an increasing number of HD genes have been identified, the association of HD and PL has been identified in pleiotropic genes. Both HD and PL are key factors that influence the business value of cultivated rice. It has been reported that PL is co-segregated and finely mapped with the HD locus, which demonstrates the pleiotropic effects of the underlying genes29,30. Hd129, Ghd715, and Ghd830 are pleiotropic, and prolong HD and enhance PL. However, genetic factors that affect HD and their effects on PL are not well understood. Moreover, a large of proportion of these QTLs have still not been finely mapped or cloned, and more HD genes are needed to elucidate the associated genetic interactions31,32.
Common wild rice (Oryza rufipogon Griff.) evolved many exotic genes in response to a variety of disasters and natural selection of harsh environments, and exhibits a late HD and open panicle architecture compared with cultivated rice33. Chromosome segments substitution lines (CSSLs) greatly improve the accuracy of gene or QTL mapping by eliminating the influence of genetic backgrounds. Thus, CSSLs have been used to obtain many cloned and fine-mapped QTLs, because they represent ideal material for genetic analysis and gene fine mapping14,34,35,36,37,38,39. Numbers of QTLs from wild rice species were reported and some alleles increase PL40,41.
OsPRR37, which is responsible for the Hd2/qDTH7-2 QTL, was reported as a major effect QTL that control photoperiod sensitivity in rice42. The genes homologous to OsPRR37 in barley, wheat, and sorghum have been well studied with regard to how they impact photoperiod sensitivity43,44,45. Natural variants of OsPRR37 have also been reported that contributed to the expansion of rice cultivation to temperate and cooler regions21. However, the natural variants of promoter of OsPRR37 have remained elusive.
To explore new allele from wild rice, we constructed a set of wild rice CSSLs in the previous study. We herein report the detection and fine mapping of a QTL for HD and PL. In this study, we performed QTL analysis for HD and PL using CSSLs and advanced backcross populations. One QTL from wild rice, qHD7.2, was identified and a novel allele of OsPRR37 was fine mapped within this QTL. Expression pattern of this gene was analyzed between wild and cultivated rice, and sequence analysis among 3000 germplasm was performed to identify the origin and selection domain during rice domestication. Our study provides a new genetic resource for cultivated rice breeding and new evidence regarding the evolution of flowering genes.
Results
qHD7.2 detection using a CSSL population
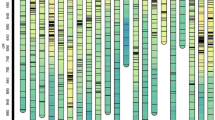
In our previous study, a set of 198 CSSLs population were developed from common wild rice as the donor parent and an elite O.sativa indica cultivar, 9311 as recurrent parent46. The HD and PL were remarkably different between the two parents; the wild rice has an open panicle phenotype and does not undergo heading under long-day conditions. The HD phenotype substantially differed within the CSSL population. The days to heading we obtained from six environments (two years, three sites) were associated with genotype based on 119 simple sequence repeat (SSR) and 62 insert/delete (InDel) polymorphic markers evenly distributed across the 12 rice chromosomes in our laboratory, several HD QTLs were identified based on a cut-off LOD score ⩾ 2.5 (Table 1). Two QTLs located, one located near RM172 on chromosome (Chr.) 7 and one located near RM19 on Chr. 12, were detected in all environments. These loci showed an increasing effect on HD; this indicates that these two QTLs were stably detected, and they could improve accuracy and efficiency of future work. In particular, the QTL near RM172 identified under the Beijing conditions had the highest LOD value (14.6) and explained 27.2% of the variance, which indicated that this QTL (named qHD7.2) is likely a main effect QTL. One CSSL, CSSL39, which flowers the latest of all CSSLs under natural long-day (NLD) conditions in Beijing (116.4°E/39.9°N, day length > 15 h) and carries the qHD7.2, was selected for advanced study. The CSSL39 genotypes were shown in Fig. 1. Seven substituted segments from wild rice were detected in the whole CSSL39 genome (Fig. 1). The markers associated with substituted segments included RM236 on Chr. 2; RM164, Indel 5-10 on Chr. 5; RM30 on Chr. 6; RM172 on Chr. 7; RM152 on Chr. 8; and RM171 on Chr. 10.

Graphical genotypes of CSSL39. Black bars indicate chromosomes of wild rice. The number of Chromosome was marked above each chromosome, the SSR markers were showed at the right of each substitution segment.
Phenotypic characterisation of CSSL39, 9311, and the F1 generation of CSSL39/9311
Compared with the recurrent parent 9311, CSSL39 exhibited a 24-d delay under NLD conditions in Beijing, 14-d delay in Nanjing, and 7-d delay under natural short-day (NSD) conditions in Sanya (110.0°E/18.5°N, day length <12 h), and the HD of F1 progeny fell in between those of the two parents (Fig. 2A,B). Additionally, we investigated CSSL39 basic agronomic traits, including thousand grain weight, grain length, grain width, plant height, flag leaf length, flag leaf width, PL, and tiller number (S-Table 1). We found that only PL was significantly shorter compared with 9311; CSSL39 PL was 5.88 cm shorter under NLD conditions and 3.58 cm shorter under NSD conditions compared with that of 9311, the F1 generation also exhibited a moderate PL (Fig. 2C,D). Moreover, we counted of the number of mature CSSL39 and 9311 seeds based on seed coat colour; the results showed that approximately 88.9% of 9311 grains but only 25% of CSSL39 grains reached maturity under NLD conditions. However, there were no significant differences in maturity between CSSL39 and 9311 under NSD conditions (S-Fig. 1), which indicated that 9311 is reproductively better adapted to the NLD conditions.

Phenotypic characterisation of CSSL39, 9311, and the F1 generation of CSSL39/9311. (A) Heading date phenotypes of CSSL39, F1, and 9311. Photos were taken under natural long-day (NLD) conditions in Beijing. (B) Heading date comparison between CSSL39 and 9311 in three environments. (C) Panicle length phenotypes of CSSL39, 9311, and F1 under NLD conditions in Beijing. (D) Panicle length comparison between CSSL39 and 9311 in three environments. P values were obtained in two-tailed t tests.
Genetic analysis and fine mapping of qHD7.2 using the CSSL39/9311 F2 population
An F2 population of CSSL39/9311 was constructed for genetic analysis and fine mapping of qHD7.2. The frequency distribution for HD and PL in the F2 segregating generation exhibited a typical normal distribution. A wide range of phenotypes presented, from 105 d to 122 d for HD and 17.2 cm to 24.7 cm for PL of 1024 individuals from Sanya, and 112 d to 146 d for HD and 16.6 cm to 29.4 cm for PL of 846 individuals from Beijing. For HD and PL, most of the F2 segregating individuals are midway between those of the two parents. Meanwhile, the existence of two-way transgressive separation was found in PL (Fig. 3).

Frequency distribution of heading date and panicle length in CSSL39/9311 F2 populations under two photoperiodic conditions (NLD and NSD). Black arrows show average heading dates and panicle lengths of CSSL39 and 9311.
Sixteen new InDel and SSR markers located at the substituted interval near RM172 were used to screen the parents, CSSL39 and 9311, of which 10 markers exhibited polymorphism (S-Table 2). First, the substituted segment near RM172 was narrowed to the interval between InDel7–12 to InDel7–13 (Fig. 4A). Combined with the eight SSR markers which were detected in CSSL39, we used these 18 polymorphic markers for CSSL39/9311 F2 population screening. HD and PL QTLs were detected in 1024 F2 individuals from Sanya and 846 F2 individuals from Beijing. In total, two HD and three PL QTLs were detected (Table 2). Two HD QTLs (qHD7.1 and qHD7.2), which can be stably inherited, were detected under both NLD and NSD conditions. qHD7.1 was mapped in the 522.9-kb interval of RM7601 to RM172 on Chr. 7, whereas qHD7.2 was mapped in the 101.1-kb interval of RM172 to RM22188 (Fig. 4B). They maintained the same genetic effects that prolong HD. Three PL QTLs (qPL7, qPL10.1, and qPL10.2) were detected. qPL7 was mapped in the 101.1-kb interval of RM172 to RM22188 at the same position of qHD7.2. qPL10.1 was mapped in the 568-kb interval of RM171 to RM1146 on Chr. 10, and qPL10.2 was mapped in the 69.7-kb interval of RM25723 to RM7300 on Chr. 10. Among those three QTLs, qPL10.1 was only detected under NSD conditions; qPL7 and qPL10.2 can be stably inherited under both NLD and NSD conditions. Only qPL10.2 under NSD conditions had a positive effect on elongating the panicle; the others had negative effects on shortening the panicle. Therefore, we predicted that qHD7.2 is likely a major QTL for delaying HD and shortening PL.

Fine mapping of qHD7.2. (A) The qHD7.2 locus was mapped between the markers Indel7–12 and Indel7–13 on chromosome 7. (B) Fine mapping of qHD7.2 using an F2 secondary segregation population. qHD7.2 was determined to belong to a 101.1-kb genomic region between the markers RM172 and RM22188. (C) Ten ORFs were identified between the RM172 and RM22188 markers. (D) Candidate gene ORF7 protein structure in CSSL39 and 9311. REC indicates signal receiver domain, CCT indicates CCT domain, and aa indicates amino acids.
Gene prediction and expression analysis
According to GRAMENE website (www.gramene.org/) and the Rice Annotation Project database (http://rapdb.dna.affrc.go.jp/)47, 10 open reading frames (ORFs) in the target region for qHD7.2 between RM172 and RM22188 were predicted (Fig. 4C, S-Table 3). Four of the ORF genes contain reported functional domains, and the others are hypothetical proteins. We sequenced each of the 10 annotated genes, and only ORF1 and ORF2 had no polymorphisms in the coding domain between CSSL39 and 9311 (data not shown). Among the 10 genes, ORF7 (LOC_Os07g49460) with functional information is related to HD control. LOC_Os07g49460 encodes a protein that contains a response regulator receiver domain and corresponds to the cloned HD gene OsPRR37; PRR37 was reported to show photoperiodic sensitivity and affects HD under long-day conditions21. Thus, we predicted that LOC_Os07g49460 could be a potential candidate for qHD7.2, and is hereafter named HD7.2. Furthermore, we aligned the HD7.2 coding domain sequence (CDS) with OsPRR37 of 9311 and Nipponbare (japonica). There were 10 mutations and an 8-bp deletion in the 9311 CDS compared with CSSL39; the 8-bp deletion produced premature translational termination and then led to a non-functional allele (Fig. 4D, S-Fig. 2). There were also five differences in the coding sequence between CSSL39 and Nipponbare, which led to five amino acid changes.
The HD7.2 spatial expression patterns were monitored in CSSL39. The HD7.2 expression levels of the flag leaf, second leaf, leaf cushion, leaf sheaths, stems, column, roots, and panicle of CSSL39 were detected under NLD conditions in the heading period. The data show that transcript levels of this gene differed among tissues; the highest expression level was found in the flag leaf, and the lowest was in the roots (Fig. 5A). We compared the HD7.2 expression levels between CSSL39 and 9311 in three different stages (before heading, heading period, and after heading), the transcript level was approximately two-fold higher in CSSL39 than in 9311 plants (Fig. 5B). Then, we compared the promoter region sequences that were located 2.0-kb upstream of the initiation codon between CSSL39 and 9311. The data show that there were many mutations or InDels in 9311 compared with CSSL39, and these polymorphic sites in the promoter region changed some cis-acting elements (S-Fig. 3); this indicated that functional proteins and the transcript level of HD7.2/OsPRR37 resulted in later flowering.

Expression analysis of HD7.2. (A) HD7.2 expression levels in different CSSL39 tissues. (B) Comparison of HD7.2/OsPRR37 transcript levels between CSSL39 and 9311 during three different stages.
Nucleotide diversity analyses and haplotype network for the PRR37
Using the Rice Functional Genomics-based Breeding (RFGB) Database (http://www.rmbreeding.cn/index), we aligned the PRR37 coding and promoter sequences to 2859 cultivated rice and 129 wild rice accessions. Abundant genetic variations were detected at LOC_Os07g49460 in the 2859 cultivated rice and 129 wild rice accessions, which included 44 non-synonymous single nucleotide polymorphisms (SNPs) in the coding sequence and 36 SNPs in their 2000-bp upstream promoter region. Two SNPs at position 1045 (A/C or T) and 2061 (C/T) were only found in cultivated rice; position 2061 is a synonymous SNP, whereas position 1045 is non-synonymous and corresponds to Lys/Gln (K349Q) amino acid replacement. Twenty-six haplotypes which contained more than 10 cultivated individuals or five wild rice individuals were selected, and Fig. 6A shows the network constructed with the major haplotypes for the LOC_Os07g49460 CDS; all individuals from H_1–12 were wild rice. H_1, H_2, and H_3 were closely related to japonica and H_4–12 was closely related to indica. Because the adjacent haplotypes in the network had very similar nucleotide polymorphisms (for example, H_2 and H_16 only had two SNPs at position 1045 and 2061, as mentioned above), the cultivated individuals might have evolved from the wild individuals with adjacent haplotypes. Therefore, O. rufipogon accessions H_1–3 were likely the direct progenitors of the O. sativa accessions H_16, H_18, H_21–22, and H_24–26, which are mostly japonica, and most of the indica accessions likely originated from other wild rice accessions in the network.

Coding (A) and promoter (B) region haplotype networks of LOC_Os07g49460 alleles. Circle size is proportional to sample quantity within a given haplotype, and the numbers next to the circle represent haplotype number. Lines between haplotypes represent mutational steps between alleles. Colours represent different species: green, O. rufipogon; blue, indica; red, japonica; and yellow, others.
The network of the LOC_Os07g49460 promoter sequence were also constructed using the same database; 21 haplotypes which contain more than 10 cultivated individuals or five wild rice individuals were selected (Fig. 6B). The promoter sequence of LOC_Os07g49460 allele had high diversity in both cultivated rice and wild rice. The cultivated rice haplotypes H_1 and H_12 were most closely related to the wild rice haplotypes H_13–15, H_17–18, and H_20. More than 75% of H_1 individuals were japonica, and all H_12 individuals were indica, which indicated that both indica and japonica promoter regions originated from wild rice and be paralleled domesticated. Other cultivated rice haplotypes, including H_2–3, H_6, and H_7–11, of which most were indica, were closely related to the wild rice haplotypes H_16 and H_19. This finding indicates that the promoter region of different cultivated rice originated from different wild rice species, and the promoter region was also selected during domestication.
Discussion
Previous studies showed that O. rufipogon from southern China is the ancestor of O. sativa, and many alleles in the wild species were lost during rice domestication48,49. The exploitation of novel alleles from wild rice that were lost in cultivated rice could be very important for rice breeding and evolution studies. Genetic populations played a major role in QTL detection and gene mapping. CSSLs have the potential to facilitate identification of QTLs not identified in F2 populations because of genetic background noise. In our previous study, we developed a CSSL population with a 9311 genetic background which cover the whole wild rice genome; many QTLs associated with various agronomic traits were identified using this CSSL population46. In this study, we identified one HD QTL, qHD7.2, using this CSSL population, which had the highest LOD value and has stable inheritance in all environments (Table 1). One CSSL, CSSL39, which consistently produced the latest heading date and carried the qHD7.2 but none of other HD QTLs from the donor parent, was selected for advanced study. CSSL39 exhibited a substantially shorter panicle than 9311 under NLD and NSD conditions. Because dense SSR markers were used during the development of this CSSL population, CSSL39 was not a near isogenic line, and eight substituted segments from wild rice were detected in its genome. Furthermore, an F2 population of CSSL39 and recurrent parent 9311 was constructed for fine mapping of qHD7.2. Ten polymorphic markers located at the substituted interval around RM172 on Chr. 7 were used for additional analysis of almost 3000 F2 plants. Finally, a pleiotropic QTL response for HD and PL was detected between two markers: RM172 and RM22188. Theoretically, an isogenic qHD7.2 line and F3 recombinants should be constructed to confirm our results. However, a previously cloned HD gene, Os07g49460 (OsPRR37), was found in this interval, and an 8-bp deletion led to a nonfunctional allele. Although another PL QTL was also detected on Chr. 10, we designed InDel primers for HD7.2 detection in F2 and F3 populations, and all individuals with the wild rice allele showed later HDs and shorter PLs (data not shown). Therefore, we can exclude other intervals, and it can be deduced that HD7.2 is the target gene involved in shaping HD and PL phenotypes. These findings also illustrate that these CSSLs are suitable and efficient for fine mapping.
Pseudo-response regulators (PRRs) have been reported to be important circadian-clock components in Arabidopsis and rice42,50. The regulatory roles of the PRR37 orthologues in growth and development diverged among species; for example, the Arabidopsis prr7 loss-of-function mutants flower slightly later under inductive long-day conditions, but rice prr37-knockout mutants flower early in non-inductive long-day conditions21,42. One member of rice PRR gene family, OsPRR37 is responsible for EH7-2/Hd2, which is the major effect QTL that controls photoperiod sensitivity in rice. OsPRR37 down-regulates Hd3a expression to suppress flowering under long-day conditions, and the natural variation in OsPRR37 regulates HD and contributes to rice cultivation at a wide range of latitudes21. Some homologous genes of OsPRR37 in barley, wheat, and sorghum have also been well documented to regulate flowering time43,44,45. In this study, the PRR37 allele from wild rice also substantially suppresses flowering under NLD conditions. Previous studies reported that PRR37 also affects other important agronomic traits such as plant height and spikelets per panicle42,43,44,45. However, no reports previously found that PRR37 affects PL, so we predicted that HD7.2 is likely a novel allele of PRR37 that delays flowering and shortens PL under long-day conditions.
Mapping of QTLs for HD and yield component traits in rice has resulted in remarkable progress in elucidating the genetic basis that underlies the natural variation of these traits. Major QTLs for HD and yield component traits have shown a common association between delayed heading and increased yield, such as Ghd715,51, DTH8/Ghd8/qHY-8/LH852,53,54,55, Hd129, and Ghd7.156. PL is a major grain yield component trait; in this study, HD7.2 delayed HD but shortened PL, potentially because HD7.2 from wild rice controls the physiological process of panicle development. Our data indicated that HD7.2 is likely a novel allele of PRR37 that has a different function. Further studies, such as a transgenic experiment of HD7.2, should be performed to understand the exact function of this gene.
There were many nucleotide changes in both coding and promoter sequences of HD7.2 between the two parents, and the expression level in CSSL39 was higher than that in 9311 during each period. In the 9311 coding sequences, an 8-bp deletion produced premature translational termination. In the promoter sequences, the changes led to cis-factor element differences between CSSL39 and 9311; these data indicated that the CSSL39 phenotype changes were caused by the changes of HD7.2 transcript level and protein function. Nucleotide diversity and network analysis of PRR37 were implemented using more than 3000 cultivated and wild rice accessions. The data showed that this gene originated from multiple wild rice accessions, which is consistent with previous reports that japonica and indica evolved from multiple ancestral populations48. Furthermore, our result showed that the promoter region also originated from wild rice, and substantial natural variation was found in rice landraces. Koo et al.21 reported that natural variation of the PRR37 protein contributed to the wide range of latitudes in which cultivated rice can be grown; usually, japonica, which is distributed in high-latitude regions, has the non-functional allele21. In this study, some landraces had functional alleles, but the promoter was low-expression type, so they still can flowering normally under long-day conditions. It can be concluded that both the HD7.2 coding and promoter regions were selected during domestication, and natural variation in the PRR37 promoter region also contributed to the widespread distribution of cultivated rice. Our study could provide a novel understanding of the rice OsPRR37 gene and rice flowering regulation networks, and provides additional evidence regarding the evolution of this gene in rice domestication.
Materials and Methods
Plant materials and growth conditions
A set of 198 CSSLs produced from common wild rice (O. rufipogon) as the donor and an elite indica variety, 9311, as the recurrent parent was developed in our laboratory as previously reported42. Each CSSL was genotyped using 313 polymorphic SSR markers evenly distributed across the 12 rice chromosomes. In this study, the CSSL population was employed for QTL mapping of HD. The genotypes of each individual were surveyed by SSR analysis; among them, one line, CSSL39, was selected as the starting material for the present study. CSSL39 was backcrossed with 9311, and the resultant F1 was self-crossed to produce F2 seeds.
The 198 CSSLs and recurrent parent 9311 were grown in six environments (two years × three locations) (S-Table 4) using the randomised complete block design with two replications. Each plot consisted of rows with 10 plants. Forty plants of each genotype in each plot were planted with a 10 × 25-cm spacing. Crop management, and disease and insect pest control were performed as locally recommended. The F2 CSSL39/9311 population was planted in Beijing from May–Oct 2016 and Sanya from Dec 2015–May 2016.
Phenotype investigation
The mean value of 10 representative individual plants in the middle of the entry plot were selected for the CSSLs and 9311. HD was measured on a single-plant basis. Days to heading for each individual were scored when the first panicle (2-cm-long) emerged. PL is the length from the panicle neck to tip of the main panicle, but does not include awn length. Seed maturity percentages were measured from two panicles sampled 60 d after 9311 heading under NLD and NSD conditions, which was determined by yellow pigmentation of the seed coat.
QTL analysis and predicted candidate gene
Initially, a total of 191 polymorphic SSR markers selected from a public database (Rice Genome Research Program 2007) were employed to construct the linkage map. To construct a high-density linkage map for fine mapping of the QTL, new InDel markers that cover the target QTL region were developed. The Nipponbare and 9311 target sequences were obtained from publicly available rice genome sequence data to develop InDel markers. Primers were designed based on InDel sequences using Primer Premier 5.0. All primer pairs flanking SSRs or InDels were designed using the following parameters: 18–25 nucleotides in length, absence of secondary structure, a GC content of approximately 50%, and a melting temperature around 55 °C. SSR and InDel marker primers were synthesised by Shanghai Invitrogen Biotechnology Company (Shanghai, China). Polymorphisms of the SSR and InDel markers between the two parents were tested by PCR. DNA of the samples was extracted from fresh leaves at the seedling stage by employing the CTAB method. PCR amplification consisted of a denaturing step of 5 min at 95 °C; followed by 33 cycles of 30 s at 94 °C, 30 s at 56 °C, and 30 s at 72 °C; finally, 10 min at 72 °C. Amplifications were separated by 6% denatured polyacrylamide gel electrophoresis and visualised by silver staining.
QTL analysis was conducted by combined the genotype with phenotype of CSSLs and secondary separation population using QTL IciMapping57. Mapping standard was identified as LOD⩾2.5, because a QTL exists when LOD ⩾2.5. Putative genes in the qHD7.2 region were predicted by referring to the Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/). Total RNA was extracted from leaves of the two parents using Trizol reagent (Invitrogen, CA, USA) and reversely transcribed into cDNA using a Reverse Transcription Kit (TaKaRa, Otsu, Japan). The coding regions of the putative genes were amplified from cDNA using PFU polymerase (TaKaRa, Otsu, Japan) and sequenced by Shanghai Sangon Biotechnology Company (Shanghai, China). DNA sequence comparison between the parents was performed using the BLAST program.
Gene expression analysis
The CSSL39 plants were grown under NLD conditions for 140 d, which was before heading. The flag leaf, second leaf, leaf cushion, stems, column, roots, panicle, and leaf sheaths were harvested. All samples were harvested from the main culm of each plant. Samples from two or three different individuals were collected as biological replicates. For expression comparison, two parents were planted in the Chinese Academy of Agricultural Sciences greenhouse. Each pot in half, planting 3 pots to make each pot have CSSL39 and 9311, to ensure consistent planting conditions. Fresh leaves were collected before heading, at heading, and after heading. RNA was extracted using TRIzol Reagent (Invitrogen, CA, USA) and treated with DNase I (Invitrogen, CA, USA). cDNA was synthesised using SuperScript III Reverse Transcriptase (Invitrogen, CA, USA). Quantitative analysis of gene expression was performed with SYBR Premix Ex Taq (TaKaRa, Otsu, Japan) on an Applied Biosystems 7500 Real-time PCR System. The data were analysed using the relative quantification method.
Network and genetic diversity analyses
We collected SNP and InDel genomic variation data for the 2859 rice genomes, and established a comprehensive SNP and InDel sub-database for the Rice Functional Genomics and Breeding Database (http://www.rmbreeding.cn/snp3k)58. This sub-database is a global resource that contains tools such as a polymorphism information retrieval function, genome browser visualization system, and data export system for specific genomic regions. All the SNPs located in the promoter and CDS regions were extracted based on the genome gff3 annotation. Haplotype analysis was performed using Perl scripts, and only non-synonymous SNPs were considered. Number of haplotypes and haplotypes diversity were counted by DnaSPv5 software (http://www.ub.edu/dnasp)59 and introduced to NETWORK 5.0.0.0 programme (Fluxus technology Ltd. 2015) for haplotype networks construction.
References
Oka, H. I. Origin of cultivated rice. Vol. 14 (Elsevier, 2012).
Khush, G. S. Origin, dispersal, cultivation and variation of rice. Oryza: From Molecule to Plant 25–34 (Springer, 1997).
Izawa, T. Adaptation of flowering-time by natural and artificial selection in Arabidopsis and rice. J Exp Bot 58, 3091–3097 (2007).
Meyer, R. S. & Purugganan, M. D. Evolution of crop species: genetics of domestication and diversification. Nat Rev Genet 14, 840–852 (2013).
Monaco, M. K. et al. Gramene 2013: comparative plant genomics resources. Nucleic Acids Res 42, D1193–D1199 (2014).
Kojima, S. et al. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol 43, 1096–1105 (2002).
Komiya, R., Yokoi, S. & Shimamoto, K. A gene network for long-day flowering activates RFT1 encoding a mobile flowering signal in rice. Development 136, 3443–3450 (2009).
Yano, M. et al. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12, 2473–2483 (2000).
Doi, K. et al. Ehd1, a B-type response regulator in rice, confers short-day promotion of flowering and controls FT-like gene expression independently of Hd1. Gene Dev 18, 926–936 (2004).
Zhao, J. et al. OsELF3-1, an ortholog of arabidopsis EARLY FLOWERING 3, regulates rice circadian rhythm and photoperiodic flowering. PLoS One 7, e43705 (2012).
Hayama, R. et al. Adaptation of photoperiodic control pathways produces short-day flowering in rice. Nature 422, 719–722 (2003).
Andrés, F. et al. Analysis of Photoperiod Sensitivity5 sheds light on the role of phytochromes in photoperiodic flowering in rice. Plant Physiol 151, 681–690 (2009).
Ishikawa, R. et al. Phytochrome B regulates Heading date 1 (Hd1)-mediated expression of rice florigen Hd3a and critical day length in rice. Mol Gen Genet 285, 461–470 (2011).
Takahashi, Y., Shomura, A., Sasaki, T. & Yano, M. Hd6, a rice quantitative trait locus involved in photoperiod sensitivity, encodes the α subunit of protein kinase CK2. Proc Natl Acad Sci USA 98, 7922–7927 (2001).
Xue, W. et al. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat Genet 40, 761–767 (2008).
Matsubara, K. et al. Ehd3, encoding a plant homeodomain finger-containing protein, is a critical promoter of rice flowering. Plant J 66, 603–612 (2011).
Peng, L. T. et al. Ectopic expression of OsLFL1 in rice represses Ehd1 by binding on its promoter. Biochem Bioph Res Co 360, 251–256 (2007).
Dai, C. & Xue, H. W. Rice early flowering1, a CKI, phosphorylates della protein SLR1 to negatively regulate gibberellin signalling. EMBO J 29, 1916–1927 (2010).
Wu, C. et al. RID1, encoding a Cys2/His2-type zinc finger transcription factor, acts as a master switch from vegetative to floral development in rice. Proc Natl Acad Sci USA 105, 12915–12920 (2008).
Song, L. K. et al. OsMADS51 is a short-day flowering promoter that functions upstream of Ehd1, OsMADS14, and Hd3a. Plant Physiol 145, 1484–1494 (2007).
Koo, B. H. et al. Natural variation in OsPRR37 regulates heading date and contributes to rice cultivation at a wide range of latitudes. Mol Plant 6, 1877–1888 (2013).
Kim, S. K. et al. OsCO3, a CONSTANS-LIKE gene, controls flowering by negatively regulating the expression of FT-like genes under SD conditions in rice. Planta 228, 355–365 (2008).
Wu, W. et al. Association of functional nucleotide polymorphisms at DTH2 with the northward expansion of rice cultivation in Asia. Proc Natl Acad Sci USA 110, 2775–2780 (2013).
Li, D. et al. Functional characterization of rice OsDof12. Planta 229, 1159–1169 (2009).
Takahashi, Y. et al. Variations in Hd1 proteins, Hd3a promoters, and Ehd1 expression levels contribute to diversity of flowering time in cultivated rice. Proc Natl Acad Sci USA 106, 4555–4560 (2009).
Liu, T., Shao, D., Kovi, M. R. & Xing, Y. Mapping and validation of quantitative trait loci for spikelets per panicle and 1,000-grain weight in rice (Oryza sativa L.). Theor Appl Genet 120, 933–942 (2010).
Huang, X. et al. Natural variation at the DEP1 locus enhances grain yield in rice. Nat Genet 41, 494–497 (2009).
Li, S. et al. Short panicle1 encodes a putative PTR family transporter and determines rice panicle size. Plant J 58, 592–605 (2009).
Zhang, Z. H. et al. Pleiotropism of the photoperiod-insensitive allele of Hd1 on heading date, plant height and yield traits in rice. PLoS One 7, e52538 (2012).
Yan, W. H. et al. A major QTL, Ghd8, plays pleiotropic roles in regulating grain productivity, plant height, and heading date in rice. Mol Plant 4, 319–330 (2011).
Bai, X., Wu, B. & Xing, Y. Yield-related QTLs and Their Applications in Rice Genetic ImprovementF. J Integ Plant Biol 54, 300–311 (2012).
Guo, L., Zhang, Z. H. & Zhuang, J. Y. Quantitative trait loci for heading date and their relationship with genetic control of yield traits in rice (Oryza sativa). Rice Sci 20, 1–12 (2013).
Rahman, M. L. et al. High-resolution mapping of two rice brown planthopper resistance genes, Bph20 (t) and Bph21 (t), originating from Oryza minuta. Theor Appl Genet 119, 1237–1246 (2009).
Song, X. J. et al. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet 39, 623–630 (2007).
Takai, T. et al. Development of chromosome segment substitution lines derived from backcross between indica donor rice cultivar’Nona bokra’and japonica recipient cultivar’Koshihikari’. Breeding Sci 57, 257–261 (2007).
Yu, B. et al. TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J 52, 891–898 (2007).
Zhou, L. et al. Fine mapping of the grain chalkiness QTL qPGWC-7 in rice (Oryza sativa L.). Theor Appl Genet 118, 581–590 (2009).
Zhou, Y. et al. Deletion in a quantitative trait gene qPE9-1 associated with panicle erectness improves plant architecture during rice domestication. Genetics 183, 315–324 (2009).
Mei, H. et al. QTLs influencing panicle size detected in two reciprocal introgressive line (IL) populations in rice (Oryza sativa L.). Theor Appl Genet 112, 648–656 (2006).
Xiao, J., Li, J., Yuan, L. & Tanksley, S. D. Identification of QTLs affecting traits of agronomic importance in a recombinant inbred population derived from a subspecific rice cross. Theor Appl Genet 92, 230–244 (1996).
Lee, S. J. et al. Identification of QTLs for domestication-related and agronomic traits in an Oryza sativa x O. rufipogon BC1F7 population. Plant Breeding 124, 209–219 (2005).
Murakami, M., Matsushika, A., Ashikari, M., Yamashino, T. & Mizuno, T. Circadian-associated rice pseudo response regulators (OsPRRs): insight into the control of flowering time. Biosci, Biotech Bioch 69, 410–414 (2005).
Turner, A., Beales, J., Faure, S., Dunford, R. P. & Laurie, D. A. The pseudo-response regulator Ppd-H1 provides adaptation to photoperiod in barley. Science 310, 1031–1034 (2005).
Beales, J. et al. A pseudo-response regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Theor Appl Genet 115, 721–733 (2007).
Murphy, R. L. et al. Coincident light and clock regulation of pseudo response regulator protein 37 (PRR37) controls photoperiodic flowering in sorghum. Proc Natl Acad Sci USA 108, 16469–16474 (2011).
Qiao, W. et al. Development and characterization of chromosome segment substitution lines derived from Oryza rufipogon in the genetic background of O. sativa spp. indica cultivar 9311. BMC Genomics 17, 580 (2016).
Sakai, H. et al. Rice Annotation Project Database (RAP-DB): an integrative and interactive database for rice genomics. Plant Cell Physiol 54, e6 (2013).
Huang, X. et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 490, 497–501 (2012).
Sun, C., Wang, X., Li, Z., Yoshimura, A. & Iwata, N. Comparison of the genetic diversity of common wild rice (Oryza rufipogon Griff.) and cultivated rice (O. sativa L.) using RFLP markers. Theor Appl Genet 102, 157–162 (2001).
Alabadı́, D. et al. Reciprocal regulation between TOC1 and LHY/CCA1 within the Arabidopsis circadian clock. Science 293, 880–883 (2001).
Weng, X. et al. Ghd7 is a central regulator for growth, development, adaptation and responses to biotic and abiotic stresses. Plant Physiol, https://doi.org/10.1104/pp.113.231308 (2014).
Yan, W. H. et al. A major QTL, Ghd8, plays pleiotropic roles in regulating grain productivity, plant height, and heading date in rice. Mol plant 4, 319–330 (2011).
Wei, X. et al. DTH8 suppresses flowering in rice, influencing plant height and yield potential simultaneously. Plant Physiol 153, 1747–1758 (2010).
Cai, H. Y. et al. Genetic and physical mapping of qHY-8, a pleiotropic QTL for heading date and yield-related traits in rice. Euphytica 184, 109–118 (2012).
Chen, J. et al. Characterization of epistatic interaction of QTLs LH8 and EH3 controlling heading date in rice. Sci Rep 4, 4263 (2014).
Yan, W. et al. Natural variation in Ghd7. 1 plays an important role in grain yield and adaptation in rice. Cell Res 23, 969 (2013).
Meng, L. et al. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. The Crop Journal 3, 269–283 (2015).
Zheng, T. Q. et al. Rice functional genomics and breeding database (RFGB): 3K-rice SNP and InDel sub-database. Chinese Science Bulletin 60(4), 367–371 (2015).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11), 1451–1452 (2009).
Acknowledgements
This research was supported by The National Key Research and Development Program of China (No. 2016YFD0100101) to Weihua Qiao and by a grant from the National Natural Science Foundation of China (No. 31471471) to Weihua Qiao. We thank Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Author information
Authors and Affiliations
Contributions
Q.W. and L.J., X.R. performed the experiments and wrote the manuscript. W.C. performed haplotype network analysis. Q.L. analysed the phenotypic data. D.Y., Z.L., C.Y., and Z.L. all contributed to PCR genotyping and gene expression analysis. Z.X. performed QTL analysis. W.W. provided haplotype data and done data analysis. Q.Y. designed the experiment. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jing, L., Rui, X., Chunchao, W. et al. A heading date QTL, qHD7.2, from wild rice (Oryza rufipogon) delays flowering and shortens panicle length under long-day conditions. Sci Rep 8, 2928 (2018). https://doi.org/10.1038/s41598-018-21330-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21330-z
This article is cited by
-
Can the Wild Perennial, Rhizomatous Rice Species Oryza longistaminata be a Candidate for De Novo Domestication?
Rice (2023)
-
OsDPE2 Regulates Rice Panicle Morphogenesis by Modulating the Content of Starch
Rice (2023)
-
Fantastic genes: where and how to find them? Exploiting rice genetic resources for the improvement of yield, tolerance, and resistance to a wide array of stresses in rice
Functional & Integrative Genomics (2023)
-
Genome-wide association studies of yield-related traits in high-latitude japonica rice
BMC Genomic Data (2021)
-
A functional chromogen gene C from wild rice is involved in a different anthocyanin biosynthesis pathway in indica and japonica
Theoretical and Applied Genetics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
