
Abstract
Genetic circuit-based biosensors are useful in detecting target metabolites or in vivo enzymes using transcription factors (Tx) as a molecular switch to express reporter signals, such as cellular fluorescence and antibiotic resistance. Herein, a phenol-detecting Tx (DmpR) was employed as a critical tool for enzyme engineering, specifically for the rapid analysis of numerous mutants with multiple mutations at the active site of tryptophan-indole lyase (TIL, EC 4.1.99.1). Cellular fluorescence was monitored cell-by-cell using flow cytometry to detect the creation of phenolic compounds by a new tyrosine-phenol-lyase (TPL, EC 4.1.99.2). In the TIL scaffold, target amino acids near the indole ring (Asp137, Phe304, Val394, Ile396 and His463) were mutated randomly to construct a large diversity of specificity variations. Collection of candidate positives by cell sorting using flow cytometry and subsequent shuffling of beneficial mutations identified a critical hit with four mutations (D137P, F304D, V394L, and I396R) in the TIL sequence. The variant displayed one-thirteenth the level of TPL activity, compared with native TPLs, and completely lost the original TIL activity. The findings demonstrate that hypersensitive, Tx-based biosensors could be useful critically to generate new activity from a related template, which would alleviate the current burden to high-throughput screening.
Similar content being viewed by others
Introduction
The recent progress in synthetic biology has encouraged the synthesis of custom-made enzymes and directed the rapid evolution of new specificities1,2,3. The combined use of rational designs and random mutagenesis has been the preferred choice in most enzyme engineering studies4,5, and has resulted in the generation of a large genetic diversity that can easily exceed one million by the random mutagenesis of just 5–6 residues. Therefore, the practical problem at this stage is the limited throughput of the screening used to identify positives, which is usually too slow to cover the genetic diversity. These rapid profiling and screening issues have become more important with the rise of synthetic biology, which requires the optimisation of multiple amino acids to generate custom-made activities or specificities in target structures6,7.
The first step to generate a custom-made specificity is the selection of a scaffold protein with appropriate structures. In this study, we selected tryptophan indole-lyase (TIL, EC 4.1.99.1) as the template to evolve a new tyrosine phenol-lyase (TPL, EC 4.1.99.2). These two lyases naturally share tetrameric structures catalysing α, β-elimination and β-replacement of their own substrates and use pyridoxal-5′-phosphate (PLP) as the coenzyme8,9.
Next, the selection of target amino acids is important to construct a focused library that may create a large diversity of possible functions in the template. For example, the design of loop libraries for the metallo-hydrolase active site could result in the detection of non-natural β-lactamase activity after extensive screening for antibiotic resistance10. More recently, a novel enzyme termed formolase was created from a benzaldehyde lyase through a focused library design and robotic high-throughput screening11. Specificity engineering with Citrobacter freundii TPL sought to confer TIL activity by replacing the active site residues with the conserved residues in TILs. However, despite the high structural similarity between two scaffolds, this attempt was not successful because of the lack of a sensitive and high-throughput assay to test the diversities of the probable mutations12.
In the present study, using screening based on phenol-detecting transcription factors (Tx) we tested the large diversities of a focused library with multiple combinations of five mutations near the substrate site of TIL, which were targeted by the comparison of structural homologues and related sequences. The diversity libraries were analysed using flow cytometry, which enables cell sorting based on cellular fluorescence and which is increasingly being used for this sort of analysis13,14,15. The fluorescence signal is obtained from specific Tx triggering the expression of cellular fluorescence in response to specific metabolites or in vivo enzymes.
We recently reported that a phenol-detecting Tx (DmpR) termed the genetic enzyme-screening system (GESS) can detect numerous phenol-generating enzymes when combined with designed substrates16. More than 200 enzyme species were assumed to be detectable by the combinations of the GESS technique and specific phenol-generating substrates, referring to the BRENDA database17. This report demonstrates that the GESS is very appropriate as a throughput tool to test the creation of the α, β-elimination of l-tyrosine in the TIL active site.
Results
Figure 1a depicts the genetic circuit-based strategy for the development of novel enzyme activities. Saturation mutagenesis diversified the selected residues in the TIL scaffold followed by the expression of gene libraries in the host cells harbouring the biosensor circuit, pGESS13. In the appearance of phenolic products, DmpR was activated to express fluorescent proteins in the corresponding cells, which were collected using flow cytometry. The obtained cells were regrown and the activity was determined using high-performance liquid chromatography (HPLC) or colorimetric analyses (Fig. 1a). The sensitivity of the flow cytometric screening was remarkably higher, while the error rate was reasonable, relative to the conventional analytical methods (inner panel of Fig. 1b).

Genetic enzyme-screening principle for evolutionary enzyme engineering. (a) Genetic libraries were constructed by site-saturation mutagenesis and staggered extension process. A genetic enzyme-screening system (GESS) was used to screen the constructed libraries for extracting active cells that triggered the genetic circuit to express a fluorescent reporter protein in the presence of phenol-lyase. Active variants with fluorescence were verified by HPLC or colorimetric assay. (b) Comparison of relative sensitivity for phenol detection by GESS (⦁), HPLC (◼) or colorimetric assay (▴). Inside panel shows the enlarged version ranging from 0 to 20 μM phenol. Each experiment was performed in triplicate.
Rational design and generation of Escherichia coli TIL mutant library
E. coli TIL was used as the template, in this study, to create TPL activity. This TIL shares many catalytic residues and mechanisms with TPLs (Fig. 2a). The recruitment of critical residues from analogous structures has been a preferred approach to design new activities in similar templates, while maintaining the residues necessary for the conserved function18,19. This approach was examined for TPL of Citrobacter freundii by recruiting residues from TIL of Proteus vulgaris. However, this attempt did not lead to the development of TIL activity in the TPL template12.

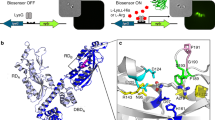
Target residues for the development of phenol-lyase activity from an indole-lyase scaffold. (a) Overlapping schemes of β-elimination of l-tyrosine and l-tryptophan catalysed by phenol-lyase (thick line and black letters) and indole-lyase (thin line and grey letters), respectively. (b) Conserved residues in the active sites of indole-lyase (upper) and phenol-lyase (lower) within a distance of 6 Å from the substrate. The y-axis represents the conservation of residues in each of 50 enzymes from the NCBI database. Asterisks indicate the characteristic residues conserved differently between indole-lyase and phenol-lyase. (c) Structural representation of five characteristic residues (Asp137, Phe304, Val394, Ile396, and His463) in tryptophan indole-lyase from Escherichia coli (PDB ID: 2C44).
In recognition of the prevalence of the genetic biosensor technology, we designed a wide diversity of mutants by selecting multiple residues to randomize within 6 Å from the substrate, referring to the conservation statistics (Fig. 2b) and three-dimensional structures of TIL (Fig. 2c). The conservation was compared with 50 sequences from the NCBI database and represented using a WebLogo diagram (Fig. 2b)20. The WebLogo diagram of the two lyases showed mostly analogous profiles, excluding five residues conserved differently in the two active sites. Asp137, Phe304, Val394, Ile396, and His463 were conserved as Thr124, Met288, Met379, Arg381, and Phe448 in the phenol-lyases, respectively (Fig. 2b). These five amino acids at the active site of E. coli TIL were selected as the target residues for saturation mutagenesis to induce promiscuity in substrate specificity. The library size was approximately 6 × 106. The focused mutation was confirmed by sequencing 30 randomly selected mutants.
Screening of E. coli TIL mutant library
The GESS-based screening procedure involving flow cytometry is summarised in Table 1. Three screening rounds were performed to identify a final TIL variant with significant TPL activity. The histogram of the 1st mutant library showed eighty-one putative hits (Fig. 3a), which were grown using solid and liquid media to confirm the increase in fluorescence. Among them, twenty-one hits showed stronger fluorescence signals than the control cells. However, when analysed by HPLC to detect phenol, none of the twenty-one hits showed a significant phenol peak (Fig. 3c; chromatogram for the 1st library hit). This was probably because the HPLC analysis was not sufficiently sensitive compared to the marginal activity developed in the TIL template. Therefore, the 2nd library was constructed (library size approximately 6 × 106) to collect the beneficial mutations among the eighty-eight mutations present in the twenty-one clones from the 1st library. Eight active variants were isolated by the flow cytometry analysis (Fig. 3a, which depicts a histogram of the 2nd library), followed by colony fluorescence analysis and HPLC. In the HPLC analysis, we identified a 2R11 clone with a significant phenol peak (Fig. 3c, which depicts a chromatogram for the 2nd library hit). The DNA sequence analysis of the 2R11 variant revealed that it harboured six mutations derived from three primary isolates: D137P and F304D from 1R123, V394L and I396R from 1R21 and H463S from 1R157 (Table 1). An unexpected mutation was identified outside the active site, I216M, in the 2nd library. This may have happened during the DNA shuffling process as random mutations can occur at the rate of 1–2 nt per kb21. To eliminate detrimental or null mutations in the 2R11 variant, the 3rd library was generated using new DNA shuffling using a 1:1 mixture of the 2R11 variant and WT TIL as the templates. Thirty-two clones with high signals were selected from this shuffling library and activity was confirmed using the colorimetric assay (Fig. 3b). Seven hits with activities higher than that of the 2R11 variant were subjected to sequence analysis and three common mutations (F304D, V394L, and I396R) and the mutation D137P were observed in six of the seven hits (Fig. 3d and Table 1). Finally, we selected the 3R3 variant with D137P, F304D, V394L, and I396R as the template for further analyses.

High-throughput screening and development of new phenol-lyases from indole-lyase mutant libraries. (a) Fluorescence histograms of the 1st and 2nd mutant libraries obtained using flow cytometry. Cells were grown in M9 broth with 1 mM L-tyrosine. (b) Fluorescence image of the 3rd mutant library on M9 agar plate containing l-tyrosine. White arrow indicates the 3R3 variant exhibiting substantial phenol-lyase activity. (c) HPLC analysis of products formed by the most beneficial indole-lyase variant sorted from each library. Citrobacter freundii phenol-lyase served as the positive control enzyme. (d) DNA shuffling of 2R11 phenol-lyase and wild-type indole-lyase. Seven hits from DNA shuffling were analysed using WebLogo to determine the frequency of each mutation. Wild-type indole-lyase residues are shown using black letters, while mutated residues are shown using red letters.
Characterisation of the new phenol-lyase, 3R3 variant
The 3R3 variant, which differed in four amino acids from E. coli TIL, was expressed in E. coli JM109 (DE3) ΔtnaA strain, purified to homogeneity and characterised for enzymatic properties (Fig. 4a). The maximum activity of the 3R3 variant for the α, β-elimination of l-tyrosine was observed at 50 °C and pH 8.0 (Fig. 4b,c), which was close to that of native TPLs22,23,24. The 3R3 variant displayed a 13-fold lower catalytic efficiency (k cat /K M = 0.58 ± 0.05) than that of the wild type (WT) C. freundii TPL and 5-fold less l-tyrosine affinity (K M = 1.18 ± 0.09) (Table 2). Specificity data of 3R3 to other substrates are summarised in Table 3. The 3R3 variant hydrolysed l-tyrosine, l-3,4-dihydroxyphenylalanine, l-serine, and S-ethyl- l-cysteine, but did not hydrolyse the original substrate, l-tryptophan.

Characterization of synthesised phenol-lyase. (a) SDS-PAGE. M: marker; Lane 1: E. coli cell extracts expressing the 3R3 variant; Lane 2: purified 3R3 enzyme. (b) Effects of temperature on 3R3 phenol-lyase. (c) Effect of pH on 3R3 phenol-lyase. All experiments were performed in duplicate.
Significance of each mutation in 3R3 phenol-lyase
In the 3R3 variant, the mutations D137P, F304D, V394L, and I396R were reverted to analyse their effects on the new phenol-lyase activity. I396R was not altered because it was presumed to perform the essential function of the base catalyst corresponding to Arg381 in the catalytic site of C. freundii TPL12. Very interestingly, any mutation of the four resides to the conserved amino acids at TIL or TPL abrogated the new phenol-lyase activity, implying that 3R3 activity was closely dependent on all the mutations that occurred (Fig. 2b). Additionally, H463 was not critical to activity, with a 45% reduction in the phenol-lyase activity in the HPLC analysis, explaining the elimination of H463S during the evolution of 3R3. As expected, no combination of single mutations could explain the development of the new enzyme activity (Fig. 5).

Effect of each mutation on 3R3 phenol-lyase activity. Relative activity of site-directed mutants of 3R3 phenol-lyase. Phenol-lyase activity was monitored for 1 h in 50 mM potassium phosphate buffer containing 50 µM pyridoxal 5-phosphate and 1 mM L-tyrosine. The concentration of generated phenol was examined using a colorimetric method. All experiments were repeated twice.
Discussion
High-throughput technologies for quantifying enzyme activities in large mutant libraries have always been a serious concern for enzyme engineering. Here, we introduce a broadly applicable high throughput screening system in conjunction with rational library design as an attempt to create new catalytic activity using a similar scaffold. The library of multiple mutations was expressed in E. coli harbouring the genetic biosensor circuit and subjected to flow cytometry. The three screening rounds enriched synergistic mutations and minimised epistatic interactions. The screening process was highly effective in isolating the early hits from numerous genetic variants owing to its extremely high sensitivity and throughput. The early hits showed no significant phenol peaks in the HPLC analysis (Fig. 3a–c). However, they could be bred using the high throughput screening that detects marginal levels of the activities in the genetic library.
The catalytic efficiency of the 3R3 variant of E. coli TIL was comparable to that of WT C. freundii TPL (7.7% in k cat /K M , Table 2), while the amino acid sequence of 3R3 differed in only four residues. First, Asp137 in E. coli TIL changed to Pro in 3R3 (Fig. 4a). Asp137 is important in the β-elimination of l-tryptophan25, while it is conserved as Thr124 in most TPLs26. Pro137 in the active site of 3R3 may possibly contribute to the proper orientation of substrates. The replacement of Pro137 with Thr in 3R3 resulted in the complete loss of TPL activity (Figs 5 and S1a). Second, Phe304 changed to Asp in 3R3 (Fig. S1b). Phe304 is involved in π-π stacking interactions with the substrate of E. coli TIL, l-tryptophan. Moreover, the position is related to a conformational change between the open and closed forms, and is conserved as Met288 in TPL27. However, the replacement of Asp304 in 3R3 with Met resulted in no detectable TPL activity (Fig. 5). Third, Val394 changed to Leu394 in 3R3 (Fig. S1c). When Leu394 changed to the consensus residue in TILs (Val) or consensus residue in native TPLs (Met), the 3R3 variant lost most of its activity and negligible levels of phenol were produced, suggesting that Leu394 was not replaceable in 3R3. Fourth, Ile396 changed to Arg in 3R3 (Fig. S1d). The residue at this position is related to Arg381 in WT TPL, which performs an essential function in β-elimination as the base catalyst12. Arg381 is critical also for the specific binding of substrate at the active site27.
In addition to the mutational studies, the 3R3 variant was subjected to molecular modelling and ligand docking simulation to determine how the α, β-elimination of l-tyrosine occurred (Fig. 6). In the energy-minimized models of 3R3 and E. coli TIL, the remarkable point was the reduced distance of 3.72 Å between the catalytic hydroxyl group of Tyr74 and Cγ of the docked l-tyrosine, whereas it was approximately 7.38 Å in the WT model (Fig. 6). Although the free energy of docking with l-tyrosine did not differ significantly between the WT and 3R3 (Table 4), the proximate distance of catalytic residues can be critical to the proper β-elimination reaction of phenol-lyase. When we performed the docking simulation of l-tryptophan into the WT and the 3R3, the docked ligand did not adequately rotate in the active site of the 3R3, whereas the WT displayed proper orientation of docked ligand, which could have been the reason for the loss of the original activity (Fig. S2).

Homology modelling of wild-type indole-lyase and 3R3 phenol-lyase. Both modelled structures were generated using molecular dynamics and docking simulations. The docked ligand and Tyr74 are represented as stick and line, whereas the 4 mutated residues are represented as balls and sticks, respectively. The distance between Tyr74 and Cγ in the substrate is marked by a green line. The new indole-lyase has a more suitable orientation of the docked ligand regarding atomic distances between the substrate and active site residues.
The diversification of five residues was sufficient to create a new phenol-lyase from a related enzyme. The new enzyme comprised unique active site residues to constitute a new TPL activity, which was not replaceable by any residues of the native enzymes. During the test of the 2nd library, an unexpected mutation, I216M, was detected outside the active site (Table 1). This indicates the occurrence of some random mutations during DNA shuffling, which is a recognised event21. However, this mutation disappeared automatically during the 3rd DNA shuffling and the high throughput screening. Presumably, the 3R3 variant can be improved further in its catalytic activity after an extensive random mutagenesis or design of a new focused library outside of the active site28,29. However, this study focused on the usefulness of hypersensitive genetic screening to create a new active site, rather than focusing on the improvement of TPL activity.
The detection of various enzyme activities using the present biosensor suggests that this method is useful for other activities as well, with the provision of an appropriate substrate with a phenolic group. The limiting factor for this approach is the design of appropriate substrates for the production of phenols and the rational design of the diversifying library from related templates. The approach could also be useful to graft new functions in native cells by merely introducing a few mutations into the homologous gene in the chromosome using precision genome engineering techniques.
This study supports the notion that desired mutations accumulate and deleterious mutations are eliminated rapidly during flow cytometric analysis, favouring changes in the substrate specificity of enzymes. The rapid development of new activity may importantly contribute to biological process development and greater understanding of the relationship between enzyme structure and function.
Methods
Materials
All reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Restriction endonucleases and T4 DNA ligase were purchased from New England Biolabs (Beverly, MA, USA) and DNA polymerase was obtained from Toyobo (Osaka, Japan). All oligonucleotides were synthesized by Bioneer Co. (Daejeon, Republic of Korea) (Table S1). DNA preparation and manipulation involved standard molecular biological protocols. A miniprep kit (Qiagen, Valencia, CA, USA) was used for plasmid DNA isolation and DNA extraction. The plasmid pGESSv4, consisting of the dmpR transcriptional activator, the Pdmp promoter from Pseudomonas putida KCTC 1452, and the egfp gene as the reporter, was used as a genetic circuit system13. The genetic circuit was designed to express enhanced green fluorescent protein (EGFP) upon the release of phenol from l-tyrosine through the catalytic activity of an intracellular phenol-lyase. The tnaA gene of the E. coli host was deleted using a lambda red recombination method30 to avoid any interference by endogenous indole-lyase. A kanamycin-resistance cassette was prepared from pKD13 using PCR primers homologous to E. coli tnaA regions and removed using pCP2031.
Construction of mutant libraries of indole-lyase
The gene encoding E. coli K-12 TIL was cloned into the plasmid pSHCE, which was derived from pHCEIIB (Takara Bio, Shiga, Japan) by replacing the p15A origin of replication from pACYC184, resulting in the generation of pSHCE-Trpase. For constructing the 1st library, the gene encoding E. coli TIL in pSHCE-Trpase was amplified by PCR with degenerate primers to diversify target residues (Table S1). The amplified DNA was cloned into pSHCE and the resulting plasmids were transformed into E. coli DH5α cells harbouring pGESSv4 to generate the 1st mutant library. All transformants were stored at −80 °C in 15% glycerol solution until daily screening using a flow cytometer. The 2nd library was constructed by DNA shuffling with the Staggered Extension Process (StEP) to recombine beneficial mutations obtained from the 1st library. An equimolar mixture of plasmids selected from the 1st library was amplified by PCR and 2 ng of this mixture was used as the template for staggered extension PCR21. The amplified DNA was cloned into the plasmid pRSF (Novagen, Darmstadt, Germany) and the resulting recombinant plasmids were transformed into E. coli EPI300 (DE3) harbouring pGESSv4 to yield the 2nd library. The 3rd library was constructed with hits from the 2nd library and WT indole-lyase following the same method and was transformed into E. coli JM109 (DE3)∆tnaA cells harbouring pGESSv4.
Library screening using flow cytometry
E. coli harbouring the mutant libraries and genetic circuit were screened using a FACSAria™ Flow Cytometer Sorter (BD Biosciences, Franklin Lakes, NJ, USA). The cells were cultured in M9 medium (6.78 g/L Na2HPO4, 3 g/L KH2PO4, 1 g/L NaCl, 2 mM MgSO4 and 0.1 mM CaCl2) supplemented with 0.4% (w/v) glucose, 0.01% (w/v) thiamine, 0.1% (w/v) leucine, 10 µM PLP, 50 µg/mL ampicillin and 25 µg/mL chloramphenicol. After culturing for 15 h at 37 °C, the culture broth was diluted 1:100 with phosphate-buffered saline (PBS) for fluorescence-activated cell sorting (FACS) analysis. A blue laser source (488 nm) and an FL1 (530/30 nm) photomultiplier tube were used to analyse EGFP fluorescence. Forward and side scatter were measured to exclude debris and dead cells. In total, 107 cells were counted for each library over 30 min. Data were acquired using BD CellQuest Pro (version 4.0.2, BD Biosciences) and analysed using Flowjo software (Flowjo, Ashland, OR, USA). The top 0.1% of cells with the highest fluorescence were sorted directly into 1 mL of Luria-Bertani (LB) medium (histogram for 1st library is presented in Fig. 3a) and grown on 10 M9 agar plates containing 1 mM L-tyrosine for further screening.
Solid and liquid-phase fluorescence analyses
Imaging of fluorescent colonies was performed using an AZ100M fluorescence Multizoom microscope (Nikon, Tokyo, Japan) equipped with a GFP filter (excitation at 455–485 nm, emission at 500–545 nm). After 48 h of incubation on M9 agar plates at 37 °C, the fluorescence of the developed colonies was observed and 100 colonies displaying fluorescence higher than that of the control cells were transferred to M9 broth containing 1 mM L-tyrosine. Fluorescence intensity was measured after 15 h at 37 °C using a multi-label plate reader (PerkinElmer, Waltham, MA, USA) at excitation and emission wavelengths of 485 nm and 535 nm, respectively.
Expression and purification of enzyme variants
Genes encoding active TIL variants and WT indole-lyase were cloned into the NcoI/EcoRV restriction site of pETDuet-1 (Novagen) and expressed in E. coli JM109 (DE3) ∆tnaA. The recombinant cells were cultured in LB medium at 37 °C in a shaking incubator and isopropyl β-D-1-thiogalactopyranoside (IPTG) was added at a final concentration of 0.5 mM, when the optical density at 600 nm (OD600) reached 0.4 and the cells were further incubated for 6 h. The cells were harvested by centrifugation at 5,000 rpm for 10 min. The pellet was resuspended in lysis buffer containing 50 mM sodium monophosphate, 300 mM NaCl, 10 mM imidazole and 0.1 mM phenylmethylsulfonyl fluoride as protease inhibitor, and disrupted by sonication for 3 min on ice. The lysate was centrifuged at 15,000 rpm for 10 min to remove cell debris. The supernatant was used directly to purify the His-tagged enzymes by loading into a Profinia™ Affinity Chromatography Protein Purification System (Bio-Rad, Hercules, CA, USA). The purified enzymes were confirmed by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and protein concentrations were quantified by the Bradford method. The purified enzymes were immediately examined for biochemical and kinetic characterisation.
Enzyme assay and HPLC analysis
Phenol-lyase activity was monitored for 1 h in 50 mM potassium phosphate buffer containing 50 µM PLP and 1 mM l-tyrosine. The concentration of the generated phenol was analysed using a HPLC system (Shimadzu, Kyoto, Japan) equipped with a C18 reversed-phase column (Chemco, Coatbridge, UK) and Chromopak UV detector (detection at 268 nm). The mobile phase consisted of 50% acetonitrile and 50% water at a flow rate of 1 mL/min. Additionally, the concentration of phenol in the reaction mixture was determined using a colorimetric method in which 100 µL of the enzyme assay solution was added to 100 µL of 0.1 M NaOH, 30 µL of 0.6% (w/v) 4-aminoantipyrine and 30 µL of 0.6% (w/v) potassium persulphate, and incubated for 10 min to evaluate the colour change using a well plate reader32. One unit (U) of the enzyme with l-tyrosine as the substrate was defined as the amount required to liberate 1 μmol phenol per minute at 50 °C and pH 8.0.
Protein-structure modelling and docking simulations
The structure of WT indole-lyase is based on its X-ray crystallographic structure with the Protein Data Bank (PDB) ID, 2C44. Indole-lyase is a tetramer with four chains, namely A, B, C, and D. Two neighbouring chains (A/B or C/D dimers) are directly involved in chemical reactions. The dimer of A and B chains was obtained from PDB for initial structure building. With the dimer, short energy minimization and molecular dynamics (MD) simulation analyses were performed to relax the protein structure. The backbone atoms were constrained to their original atomic coordinates using harmonic potential. The force constant of the harmonic potential gradually decreased from 1, 0.5, and 0.0 kcal/mol as MD simulation progressed. The initial structure of the 3R3 variant was based on the structure of WT indole-lyase. For initial structure building for the 3R3 variant, common atomic positions, such as backbone, Cβ atoms and so on, from WT indole-lyase were maintained. The remaining missing atoms (D137P, F304D, V394L, and I396R) were generated using internal coordinate manipulation from Chemistry at HARvard Macromolecular Mechanics (CHARMM)33. The same protocol was used for the generation of the structure of WT indole-lyase. After MD simulations, two relaxed structures were obtained and they were used for docking simulation by AutoDock Vina with l-tyrosine and l-tryptophan as ligands34. The pocket residues for docking were identified using the pocket detection programme Pck (http://schwarz.benjamin.free.fr/Work/Pck/home.htm). With the detected pocket residues, (296 residues detected), in total 2,960 docking simulations were performed involving ten attempts of docking simulations for each pocket residue with different random seeds. The 2,960 docked ligand poses were clustered and grouped. Clustering was based on the centres of mass (COMs) of the docked ligands. The group with the lowest energy conformation was chosen for further analyses. The chemical activities of WT and mutant indole-lyase were quantified in terms of the docking energy (unit being kcal/mol) of the ligands and their orientation within the binding pocket. The appropriate orientation of tyrosine was expressed in the atomic distance between tyrosine and active site residues.
References
Pleiss, J. Protein design in metabolic engineering and synthetic biology. Current opinion in biotechnology 22, 611–617 (2011).
Copley, S. D. Toward a systems biology perspective on enzyme evolution. The Journal of biological chemistry 287, 3–10 (2012).
Chen, Z. & Zeng, A. P. Protein engineering approaches to chemical biotechnology. Current Opinion in Biotechnology 42, 198–205 (2016).
Chica, R. A., Doucet, N. & Pelletier, J. N. Semi-rational approaches to engineering enzyme activity: combining the benefits of directed evolution and rational design. Current opinion in biotechnology 16, 378–384 (2005).
Lutz, S. Beyond directed evolution-semi-rational protein engineering and design. Current Opinion in Biotechnology 21, 734–743 (2010).
Gerlt, J. A. & Babbitt, P. C. Enzyme (re)design: lessons from natural evolution and computation. Current opinion in chemical biology 13, 10–18 (2009).
Heinisch, T. & Ward, T. R. Design strategies for the creation of artificial metalloenzymes. Current opinion in chemical biology 14, 184–199 (2010).
Ku, S. Y., Yip, P. & Howell, P. L. Structure of Escherichia coli tryptophanase. Acta crystallographica. Section D, Biological crystallography 62, 814–823 (2006).
Milić, D. et al. Crystallographic snapshots of tyrosine phenol-lyase show that substrate strain plays a role in C-C bond cleavage. Journal of the American Chemical Society 133, 16468–16476 (2011).
Park, H.-S. et al. Design and evolution of new catalytic activity with an existing protein scaffold. Science (New York, N.Y.) 311, 535–538 (2006).
Siegel, J. B. et al. Computational protein design enables a novel one-carbon assimilation pathway. Proceedings of the National Academy of Sciences 112, 3704–3709 (2015).
Phillips, R. S., Demidkina, T. V. & Faleev, N. G. Structure and mechanism of tryptophan indole-lyase and tyrosine phenol-lyase. Biochimica et Biophysica Acta (BBA)—Proteins & Proteomics 1647, 167–172 (2003).
Choi, S. L. et al. Toward a generalized and high-throughput enzyme screening system based on artificial genetic circuits. ACS synthetic biology 3, 163–171 (2014).
Jha, R. K., Kern, T. L., Fox, D. T. & M Strauss, C. E. Engineering an Acinetobacter regulon for biosensing and high-throughput enzyme screening in E. coli via flow cytometry. Nucleic acids research 42, 8150–8160 (2014).
Pitzler, C. et al. A Fluorescent Hydrogel-Based Flow Cytometry High-Throughput Screening Platform for Hydrolytic Enzymes. Chemistry & Biology 570, 1733–1742 (2014).
Lee, D.-H. et al. A novel psychrophilic alkaline phosphatase from the metagenome of tidal flat sediments. BMC Biotechnology 15, 1–13 (2015).
Kim, H., Kwon, K. K., Rha, E. & Lee, S.-G. Genetic Enzyme Screening System: A Method for High-Throughput Functional Screening of Novel Enzymes from Metagenomic Libraries. https://doi.org/10.1007/8623 (2015).
Khersonsky, O. & Tawfik, D. S. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annual review of biochemistry 79, 471–505 (2010).
Furnham, N. et al. Exploring the evolution of novel enzyme functions within structurally defined protein superfamilies. PLoS computational biology 8, https://doi.org/10.1371/journal.pcbi.1002403 (2012).
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome research 14, 1188–1190 (2004).
Zhao, H., Giver, L., Shao, Z., Affholter, J. A. & Arnold, F. H. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat Biotechnol 16, 258–261 (1998).
Watanabe, T. & Snell, E. E. Reversibility of the tryptophanase reaction: synthesis of tryptophan from indole, pyruvate, and ammonia. Proceedings of the National Academy of Sciences of the United States of America 69, 1086–1090 (1972).
Lee, S.-G. et al. Inactivation of tyrosine phenol-lyase by Pictet-Spengler reaction and alleviation by T15A mutation on intertwined N-terminal arm. The FEBS journal 273, 5564–5573 (2006).
Phillips, R. S., Ghaffari, R., Dinh, P., Lima, S. & Bartlett, D. Properties of tryptophan indole-lyase from a piezophilic bacterium, Photobacterium profundum SS9. Archives of biochemistry and biophysics 506, 35–41 (2011).
Demidkina, T. V. et al. Role of aspartate-133 and histidine-458 in the mechanism of tryptophan indole-lyase from Proteus vulgaris. Biochemistry 42, 11161–11169 (2003).
Demidkina, T. V. et al. Threonine-124 and phenylalanine-448 in Citrobacter freundii tyrosine phenol-lyase are necessary for activity with L-tyrosine. Biochem. J. 363, 745–752 (2002).
Milić, D., Demidkina, T. V., Faleev, N. G., Matković-Calogović, D. & Antson, A. A. Insights into the catalytic mechanism of tyrosine phenol-lyase from X-ray structures of quinonoid intermediates. The Journal of biological chemistry 283, 29206–29214 (2008).
Jeffery, C. J., Gloss, L. M., Petsko, G. A. & Ringe, D. The role of residues outside the active site: structural basis for function of C191 mutants of Escherichia coli aspartate aminotransferase. Protein Eng. 13, 105–112 (2000).
Mendonça, L. M. F. & Marana, S. R. Single mutations outside the active site affect the substrate specificity in a β-glycosidase. Biochimica et Biophysica Acta—Proteins and Proteomics 1814, 1616–1623 (2011).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proceedings of the National Academy of Sciences of the United States of America 97, 6640–6645, https://doi.org/10.1073/pnas.120163297 (2000).
Cherepanov, P. P. & Wackernagel, W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14 (1995).
Otey, C. R. & Joern, J. M. High-throughput screen for aromatic hydroxylation. Methods in molecular biology (Clifton, N.J.) 230, 141–148 (2003).
Brooks, B. R. et al. CHARMM: The biomolecular simulation program. Journal of Computational Chemistry 30, 1545–1614 (2009).
Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry 31, 455–461 (2010).
Acknowledgements
This work was supported by grants from the Intelligent Synthetic Biology Centre of Global Frontier Project (2011-0031944), KRIBB Research Initiative Program and C1 Gas Refinery Program through the National Research Foundation of Korea (NRF2015M3D3A1A01064875).
Author information
Authors and Affiliations
Contributions
S.L. and K.K.K. designed the study; S.L. and K.J. supervised the work; S.J.K. and J.L. conducted the experiments for protein structure prediction; K.K.K., S.C. and E.R. performed the construction and screening of libraries; B.S. and S.Y. performed enzyme purification and kinetic characterization; K.K.K., S.L. and D.L. analysed the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kwon, K.K., Lee, DH., Kim, S.J. et al. Evolution of enzymes with new specificity by high-throughput screening using DmpR-based genetic circuits and multiple flow cytometry rounds. Sci Rep 8, 2659 (2018). https://doi.org/10.1038/s41598-018-20943-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20943-8
This article is cited by
-
Heme biosensor-guided in vivo pathway optimization and directed evolution for efficient biosynthesis of heme
Biotechnology for Biofuels and Bioproducts (2023)
-
An integrative approach to improving the biocatalytic reactions of whole cells expressing recombinant enzymes
World Journal of Microbiology and Biotechnology (2021)
-
Acclimation of bacterial cell state for high-throughput enzyme engineering using a DmpR-dependent transcriptional activation system
Scientific Reports (2020)
-
Current Status of Pseudomonas putida Engineering for Lignin Valorization
Biotechnology and Bioprocess Engineering (2020)
-
Genetically encoded biosensors for lignocellulose valorization
Biotechnology for Biofuels (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
