
Abstract
Genomic loci related with resistance to gall-inducing insects have not been identified in any plants. Here, association mapping was used to identify molecular markers for resistance to the gall wasp Leptocybe invasa in two Eucalyptus species. A total of 86 simple sequence repeats (SSR) markers were screened out from 839 SSRs and used for association mapping in E. grandis. By applying the mixed linear model, seven markers were identified to be associated significantly (P ≤ 0.05) with the gall wasp resistance in E. grandis, including two validated with a correction of permutation test (P ≤ 0.008). The proportion of the variance in resistance explained by a significant marker ranged from 3.3% to 37.8%. Four out of the seven significant associations in E. grandis were verified and also validated (P ≤ 0.073 in a permutation test) in E. tereticornis, with the variation explained ranging from 24.3% to 48.5%. Favourable alleles with positive effect were also mined from the significant markers in both species. These results provide insight into the genetic control of gall wasp resistance in plants and have great potential for marker-assisted selection for resistance to L. invasa in the important tree genus Eucalyptus.
Similar content being viewed by others


Introduction
There are approximately 132,930 insect species around the world that can infect plant tissues and induce tumor-like gall formation1. Gall-inducing insects (gallers) belong principally to the orders Diptera (mainly family Cecidomyiidae), Hymenoptera (mainly Cynipidae), Hemiptera and Thysanoptera1,2. Although gall tissues provide a protected nutrient-rich and favourable microenvironment to the gallers3, many of the gallers act as parasites to plants and affect adversely host growth, thereby posing serious agricultural and forestry threats4,5. For instance, the leave- or root-galling phylloxera (Daktulosphaira vitifoliae Fitch) devastated the grape (Vitis vinifera L.) production and wine industry in Europe in the 1860s and again threatened the viticulture in California in the 1980s6. Thus, resistance to galler pests has been a pivotal breeding objective in those crops subject to galling risk.
The woody plant genus Eucalyptus L′Hér. (family Myrtaceae) is almost entirely native to the Australian continent and adjacent islands7. Eucalyptus trees (eucalypts) have been cultivated worldwide for timber, fuel, pulp and paper purposes, with global plantations totaling at more than 21 million ha8. In their native range, eucalypts sustain a rich fauna of gall-inducing insects9 and are also specific hosts to several gall wasps in the family Eulophidae (Hymenoptera: Chalcidoidea), including Leptocybe invasa Fisher & La Salle (Fig. 1) which typically induces bump-shaped galls on the leaf midribs, petioles and stems of young susceptible trees10. L. invasa can cause retardation of host growth and devastating damage to eucalypt nurseries and plantations11. Outside the native range of eucalypts, L. invasa was first observed in Israel in 2000 and spread subsequently over many other regions in Africa, Europe, Asia and South America10,11,12. Biological controls have been attempted using parasitoid wasps like Quadrastichus mendeli Kim & La Salle and Selitrichodes kryceri Kim & La Salle13, however, selection of resistant or less susceptible genotypes has potential in mitigating the damage from the galler insect given the fact that variation in resistance exists among species, provenances, genotypes and/or clonal varieties in Eucalyptus10,11,12,14,15,16.

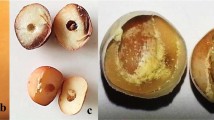
The galling insect Leptocybe invasa and its infection sympotoms on Eucalyptus. (a) L. invasa adults (~1.2 mm in length) attacking eucalypt shoots. (b) Galls on leaf and petiole (E. tereticornis), (c) galls on tender stem and branch (E. grandis) and (d) multiple sprouts (E. grandis) after L. invasa infection. Photos were taken by Huanhua Huang.
Exploration of genetic variation related with phenotypic differences will shed insights into the genetic mechanism underlying trait formation and identify valuable markers for selection in breeding schemes. In recent decades, the wealth of molecular markers developed in many plant species has enabled the genetic dissection of phenotypic traits using DNA based approaches, including quantitative trait locus (QTL) mapping, association (or linkage disequilibrium, LD) mapping (AM) and genomic selection17. QTL mapping uses bi-parental segregating populations to determine genomic regions influencing complex traits and has proved to be useful in a wide spectrum of plants18,19. Genomic selection applies breeding populations to scan genome-scale molecular data for optimal phenotypes and holds great promise for plant breeding efforts20. However, both QTL mapping and genomic selection have relatively low resolution with the causative genetic variant, and marker-gene linkage can be lost through recombination in other populations or advanced generations, thus limiting their applications in breeding and gene function studies17. In this regard, AM has been proposed as a powerful method for fine mapping because it can take full advantage of LD and historical recombinations in natural populations to identify molecular markers located within the extent of LD decay of a causal or functional genetic variant19,21,22. To date, though QTL mapping has resulted in discovery of several genomic loci in plants related with resistance to insects, such as sorghum [Sorghum bicolor (L.) Moench] to Spodoptera frugiperda J. E. Smith (Lepidoptera: Noctuidae)23 and soybean [Glycine max (L.) Merr.] to Aphis glycines Matsumura (Hemiptera: Aphididae)24, few AM studies have involved insect resistance25. Specifically, genomic loci related with galler insect resistance have not yet been reported in any plants, and the genetic mechanism underlying such a resistance remains to be clarified.
AM can be performed via genome-wide marker or candidate gene (CG) approaches. For outcrossing species that exhibit great genetic diversity and rapidly decaying LD, e.g. within about 1,500 bp on average in maize (Zea mays L.)26, a typical genome-wide AM may need tens of millions of markers to be accurately genotyped with numerous individuals, which is still challenging and costly22. Consequently, the CG approach has been widely adopted for AM work with outcrossing plants, such as maize27,28 and several forest trees17,29. However, this approach is inherently limited by the a priori choice of CGs which precludes the causal mutations located in nonidentified CGs21. Also, the trait variation explained by individual markers (usually single nucleotide polymorphism, SNP) is very low and rarely exceeds 5%30. In particular, it is impractical for those traits that no CGs have been discovered. On the other hand, genome-wide LD decay has been revealed to be substantially slower in outcrossing plants (e.g. approximately 3.7–5.7 kb with the largest LD up to 50 kb in E. grandis31) than previous estimates with CGs. More recently, with the rapid development of genomic technologies and resources, genome-wide AM has been attempted using next generation sequencing based SNPs (e.g. maize32), microarray-based SNPs [e.g. Picea glauca (Moench) Voss33] and microsatellites [or simple sequence repeats, SSR; e.g. rice (Oryza sativa L.)34, Punica granatum L.35, Theobroma cacao L.36 and Ipomoea batatas L.37].
In this study, we used a select set of SSR markers to perform AM in E. grandis Hill ex Maiden for resistance to the gall wasp L. invasa and verified the associated SSRs in E. tereticornis Smith. SSRs have been the choice of markers for AM studies in many selfing34,38,39 and outcrossing plants35,36,37. Both E. grandis and E. tereticornis are important species in terms of breeding and genomic efforts40, and E. grandis is the second tree genome (after Populus trichocarpa Torr. & Gray) to be sequenced41. Variation in L. invasa resistance has been observed in E. grandis15 and also E. tereticornis16. Low population differentiation has been identified in E. grandis by isozyme markers (GST = 0.12)42 and SSRs (FST = 0.037)43 and also in E. tereticornis by SSRs (FST = 0.012)44, suggesting a weak population structure that is ideal for AM analyses. Furthermore, though verification of association in additional population(s) is a valuable tool to demonstrate cross-population utility, only a few studies in plants have to date conducted it17. The objectives of this study were to (1) detect and verify the marker loci associated with resistance to L. invasa and (2) identify the favourable alleles for potential use in marker assisted selection in Eucalyptus. So far to our knowledge, this is the first report of mapping genomic loci associated with gall-inducing pest resistance in plants.
Materials and Methods
Plant materials
A total of 470 individual trees of E. grandis were sampled as a ‘discovery’ population from a provenance/progeny trial located at Zhaoqing City (112°27′E, 23°03′N), Guangdong Province, China. The trial was laid out following a randomized complete block design, with 32 replicates of single-tree (per family) plots at 2 × 3 m spacing. One to five trees (the first five replicates) were sampled from each of 158 open-pollinated (half-sib) families representing 16 natural seed sources (provenances) across the range of E. grandis in Australia43. A ‘verification’ population of 303 individual trees of E. tereticornis was sampled from a provenance/progeny trial located at Zhanjiang City (110°05′E, 21°16′N), Guangdong Province, China, which had been planted with four replicates of four-tree (per family) row plots in a randomized complete block design16. One to seven trees (the first two replicates) were sampled from each of 77 open-pollinated families from 11 natural provenances in Australia as described earlier16. Leaf samples were collected in July 2011 and March 2015 for E. grandis and E. tereticornis at ages of 15 and 31 months after planting, respectively. The leaves were stored at −80 °C prior to DNA extraction.
Assessment of L. invasa infestation
Natural infestation of L. invasa was assessed for the E. grandis trial at age of 15 months when the gall incidence was evident. As different infestation indices were adopted in the literature11,14,15, we employed a five-grade criterion based on the number of galls visible on a whole tree as performed similarly by Goud et al.14, namely, grade 1 = 50 and more galls, grade 2 = 10–49 galls, grade 3 = 9 and less galls, grade 4 = multiple sprouts without galls and grade 5 = no symptom.
For the E. tereticornis trial, susceptibility by L. invasa was scored previously at nine months based on the percentage of galled leaves and twigs16. The scores were then approximated to the above five-grade criterion depending on infested leaf and twig numbers assuming a mean of two galls per leaf or twig. As the number of galls is strongly positively correlated with mean severity score (based on percentage infestation/100) and proportion of plants infested12, such an approximation would provide valid estimation of the infestation grades.
Phenotypic variation in L. invasa infestation was assessed using the statistical software SAS/STAT® 8.1 (SAS Institute Inc., Cary, NC, USA). Analysis of variance (ANOVA) was conducted only for E. grandis (based on the relatively complete replicates 1−4) as that of E. tereticornis had been reported earlier for the whole trial16. Narrow-sense heritability \(({h}_{i}^{2})\) was calculated as: \({h}_{i}^{2}=1/r\times {\sigma }_{F}^{2}/({\sigma }_{F}^{2}+{\sigma }_{P}^{2}+{\sigma }_{E}^{2})\), where r is the coefficient of relationship between the individuals within families (r = 0.40 for most open-pollinated families from natural stands of Eucalyptus45), \({\sigma }_{F}^{2}\) is the among-family variance within provenances, \({\sigma }_{P}^{2}\) is the among-provenance variance, and \({\sigma }_{E}^{2}\) is the residual error variance. Standard error of h i 2 was estimated using the delta method46.
DNA extraction and SSR marker assay
Genomic DNA was extracted from leaf samples (~300 mg per sample) using a modified cetyltrimethyl ammonium bromide (CTAB) method47. DNA concentration and quality were assessed using 1.2% agarose gel electrophoresis and a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA).
A total of 839 genome-wide SSRs as used in Li et al.48 were initially tested with one E. grandis DNA sample using routine polymerase chain reaction (PCR) amplification49. Those markers (561 SSRs, 66.9%) each resulting in a single clear band in agrose gel electrophoresis were subsequently screened against two sample pools of E. grandis, namely, resistant pool (four and four samples at grades 5 and 4, respectively) and susceptible pool (eight samples at grade 1). The markers (86 SSRs distributing across the 11 main scaffolds and a small scaffold of E. grandis genome; Supplementary Fig. S1) that exhibited at least 0.20 of allelic frequency difference between the resistant and susceptible pools were finally selected out for genotyping the ‘discovery’ population of E. grandis. The SSR genotyping method followed the fluorescein-12-dUTP based procedure as described earlier49.
In addition, 25 and 12 putatively neutral genomic SSRs (Supplementary Table S1) were used for population structure analysis in E. grandis and E. tereticornis, respectively. These SSRs were previously reported to neither depart significantly from Hardy-Weinberg equilibrium (HWE; P < 0.01) nor show outlying between-population differentiation (FST) values in E. grandis43 and/or E. tereticornis44.
Marker polymorphism, linkage disequilibrium (LD) and population structure
For E. grandis, number of alleles (NA), observed heterozygosity (HO), expected heterozygosity (HE), allele size range (ASR) and polymorphic information content (PIC) per SSR marker were estimated with MSA software50. LD between the SSRs was evaluated using TASSEL 3.0 software51. The determination coefficient (r2) was used to test the LD pattern with 100,000 permutations.
STRUCTURE 2.3.4 software52 was performed to cluster individuals into a number (K = 1−16 and 1−11 for E. grandis and E. tereticornis, respectively) of genetically homogeneous sub-populations based on an admixture model with correlated allele frequencies between provenances. For each K value, the Markov Chain Monte Carlo (MCMC) sampling was replicated with 10 runs53 each following 100,000 burn-ins and 100,000 MCMC iterations. The optimal K value was determined with the highest ΔK method54 in STRUCTURE HARVESTER 0.655. The membership coefficient (Q) of each individual generated under the optimal K value was used to form the population structure Q matrix. Also, pair-wise kinship coefficients (K matrix) between individuals were estimated using SPAGeDi1-5a software56. The Q and K matrices were incorporated into the subsequent association analysis.
Association mapping and verification
A mixed linear model (MLM) was performed using TASSEL 3.0 software51 [file type option ‘Load polymorphism alignment (custom)’] for marker-trait association mapping in E. grandis and association verification in E. tereticornis. In order to avoid possible spurious associations, Q and K matrices generated above were incorporated as co-variates (Q + K method). The significant association probability was set at P ≤ 0.05. The R2 value indicated the percentage of phenotypic variance explained by the marker identified. Only markers with allele frequencies of 5% or higher were included in association analysis. Also, the significant associations were further validated with a correction of permutation test (P ≤ 0.008 and 0.073 for E. grandis and E. tereticornis, respectively). The significant markers were function annotated by BlastX search of their original sequences against NCBI non-redundant protein database (https://blast.ncbi.nlm.nih.gov/Blast.cgi) with a cutoff E-value of 10−5.
Phenotypic allele effect was estimated in comparison to the average phenotypic value of ‘null allele’ (including the rare alleles with frequency less than 5%)57. An allele of positive effect was identified as favourable allele for L. invasa resistance. The general mean of positive or negative allelic effects was calculated as the average (positive or negative) allelic effect (AAE) of a marker, and its percentage taking account of the average ‘null allele’ phenotypic effect was also calculated58.
Results and Discussion
L. invasa resistance variation
The mean value of L. invasa resistance was slightly smaller in E. grandis than E. tereticornis (Table 1). In E. grandis, ANOVA indicated nonsignificant differences in L. invasa resistance among provenances and among families within provenances (Supplementary Table S2). The h i 2 estimate (0.10 ± 0.02; Table 1) was low, especially compared to that of E. tereticornis (0.52 ± 0.50) calculated from the whole trial16. However, the h i 2 for both E. grandis and E. tereticornis may be at similar magnitude considering the relatively high value of stand error shown in E. tereticornis16.
SSR marker polymorphism, LD and population structure
A total of 1,644 alleles were detected at the 86 SSR markers selected for association mapping in the E. grandis ‘discovery’ population. Polymorphic parameters differed markedly among markers, with NA ranging from three to 60 (mean 19.1), HO from 0.1608 to 0.9893 (mean 0.6305), HE from 0.2049 to 0.9694 (mean 0.7771) and PIC from 0.1847 to 0.9673 (mean 0.7549; Supplementary Table S3). The level of LD between the 86 SSRs inferred from E. grandis was generally low, with r2 from 0 to 0.0878 (mean 0.0033) and only 87 (2.4%; P < 0.01) of the pairwise correlations showing significant LD (Fig. 2). Significant LD existed between linked and/or unlinked markers (Fig. 2).

Distribution of LD (r2 value) among 86 SSR markers in E. grandis. SSR markers and their original scaffolds are along the X-axis. Each pixel above the diagonal represents the r2 value size (color code as shown in the upper right) of an SSR marker pair, and each pixel below the diagonal represents the P value size (color code as shown in the lower right) for testing the LD.
Out-crossing plant species including eucalypts are expected to show a lower extent of LD compared to selfing plants21. The LD detected here in E. grandis is much lower than those estimated earlier in Eucalyptus. For example, Arumugasundaram et al.59 reported r2 values of 0−0.133 (mean 0.09) and 0−0.62 (mean 0.012) in 40 E. camaldulensis Dehnh. and 50 E. tereticornis trees, respectively, based on 62 SSRs, and Silva-Junior and Grattapaglia31 reported average genome-wide r2 of 0.131 in 48 E. grandis trees (two provenances) based on 21,351 SNPs. As population background can affect LD39, the lower LD level observed in this study could be mostly due to the larger size of population analysed (range-wide plant materials). Also, the extent of LD could vary with marker (genomic) loci39. Consequently, in light of a lower LD, a higher resolution of marker-trait associations can be expected.
Population structure analysis indicated that the optimal K value was determined to be two for the E. grandis ‘discovery’ population (Fig. 3a), which was in agreement with previous PCA analysis on the same population43. The 470 individuals were thus divided into two sub-populations (Fig. 3c). Similarly, the 303 individuals of the E. tereticornis ‘verification’ population were also divided into two sub-populations (Fig. 3b and d). These results corroborate the previous division of two genetically distinct clusters of natural populations in E. grandis43 and E. tereticornis44, indicating weak genetic structure among provenances for both species. Moreover, population structure can result in spurious marker-trait relations in subsequent association mapping38, and the appropriate identification of genetic structure, though weak in our cases, will help to eliminate false marker-trait associations.

Two sub-populations inferred for 470 E. grandis and 303 E. tereticornis trees based on 25 and 12 putatively neutral genomic SSR markers, respectively. Optimal K value was two as determined from ΔK change with K in (a) E. grandis and (b) E. tereticornis. Two sub-populations were then partitioned with membership coefficient (Q) for each of (c) E. grandis and (d) E. tereticornis.
Association mapping and verification
There were seven SSR markers associated in E. grandis with L. invasa resistance at the P ≤ 0.05 significance level, of which two (EUCeSSR0755 and EUCeSSR479) were validated with a correction of permutation test (P ≤ 0.008; Fig. 4, Table 2 and Supplementary Table S4). The R2 value of a significant marker ranged from 3.3% (EUCeSSR0930) to 37.8% (Embra333), with an average of 16.7%. The seven SSRs resided on scaffolds 2, 3, 6, 7, 8 and/or 5 of the E. grandis genome (Table 2). Further, four of the seven significant associations were verified and also validated in E. tereticornis (P ≤ 0.073 in permutation test; Table 2), with R2 ranging from 24.3% to 48.5% (averaging at 34.3%). All of the verified markers had consistently higher R2 in E. tereticornis. High R2 values have been demonstrated for SSR markers in other plants, e.g. the highest being 80% for yellow mosaic virus disease resistance in soybean38 and 20% for aluminum tolerance in rice34. The high R2 may be attributable to the multiple alleles of an SSR, of which the effect of each allele, usually small, can be accumulated to an extraordinarily high level.

Distribution of significance levels for the 86 SSRs tested for association with L. invasa resistance in E. grandis. SSRs and their original scaffolds are along the X-axis. P values were transformed as −log10P (Y-axis). *P ≤ 0.05; **P ≤ 0.01 with validation (P ≤ 0.008) in a correction of permutation test. Four markers (underlined) were verified to be significant in E. tereticornis.
Little is known about the genomic loci associated with response to gall wasps in plant species, and the significant markers identified here would therefore provide insight into the genetic control of insect resistance in plants. In addition to the low h i 2 estimate, multiple significantly associated markers suggest the quantitative inheritance of gall wasp resistance in Eucalyptus. Of the seven significant loci detected in E. grandis, five (Embra333, EUCeSSR0930, Embra321, EUCeSSR479 and EUCeSSR683) are homologous to known genes or predicted proteins when their original sequences were BlastX searched against the NCBI non-redundant protein database. The locus Embra333 is functionally annotated as a C2H2 zinc finger protein (Cynara cardunculus var. scolymus L.; 9e−17 and 64% in E-value and similarity, respectively). C2H2 zinc finger proteins are one of the largest transcript factor families in plants and have been found to participate in diverse signal transduction pathways and developmental processes, including pathogen defense and stress responses60. EUCeSSR0930 has homology to M-phase inducer phosphatase 3 (Anthurium amnicola Dressler; 5e−22 and 70% in E-value and similarity, respectively), a protein that can be phosphorylated and activated by Cdkl/cyclin B and leads to entry into mitosis61. Embra321 is homologous to a gene annotated as predicted U-box domain-containing protein 51-like (E. grandis; 1e−8 and 85% in E-value and similarity, respectively). Though the physiological function of U-box domain remains unclear, plant U-box proteins have been implicated as regulators of fundamental cellular processes related to signal transduction, damage responses and programmed cell death as well as defense against biotic and abiotic stresses62. EUCeSSR479 is functionally related to membrane-anchored endo-1,4-beta-glucanases (Gossypium hirsutum L.; 8e−79 and 86% in E-value and similarity, respectively), which are involved in cellulose biosynthesis in plants63. Also, genes encoding endo-1,4-beta-glucanases have been found in bacteria, fungi, nematodes and insects. In the crown gall-forming bacterium Agrobacterium tumefaciens, cellulose fibers can be produced to adhere to plant cell walls during infection64. EUCeSSR683 is a predicted proline-rich receptor-like protein kinase PERK9 (E. grandis; 3e−22 and 98% in E-value and similarity, respectively), which is expressed widely in Arabidopsis thaliana L. Heynh.65 and may act as a sensor/receptor in plants to monitor changes at cell walls during cell expansion or during exposure to abiotic/biotic stresses and then activate associated cellular responses66. However, the remaining two loci (EUCeSSR0755 and Embra345) were of unknown function. As EUCeSSR0755 is derived from an expressed sequence tag (ES589368), it may be a CG for physiological response to gall wasp infection. Nevertheless, causal gene(s) might be located in the LD region of a significant marker locus as neutral markers may represent artificial association caused by genetic hitchhiking67.
Few associations in plants have been verified in a different species though several studies have verified association results in an additional full-sib mapping population of the same species, such as yellow mosaic virus resistance associated SSRs in G. max38 and wood-property associated SSRs in Populus tomentosa Carr.68. In our study, four of the seven significant markers in E. grandis were verified in E. tereticornis, suggesting the effectiveness of these marker-trait associations across species. However, three SSR markers remained non-significant in verification analysis with E. tereticornis. Several factors are possible to affect the verification results, including species, environment, population size and phenotyping. Related species may contain different loci affecting such a complex trait as insect resistance, which could have evolved independently in different populations and habitats69. Moreover, low LD in forest trees can give rise to inconsistent marker-trait associations among genotypes even within the same species70. Also, coupled with environment and population size, phenotyping technique plays an important role in finding accurate genotype-phenotype associations25. In the present study, phenotyping of gall wasp resistance was different between the ‘discovery’ and ‘verification’ species in terms of trial site, measurement age, season, population size and phenotyping method, which could be attributable, at least in part, to those non-verified associations in E. tereticornis. As the factors mentioned above are concerned, further efforts need to carry out multiple-site experiments deploying a large amount of clonally propagated genotypes.
Mining for favourable alleles and implications for practical breeding
The alleles with positive effects are considered as favourable alleles for L. invasa resistance. Table 3 shows the first allele with the largest (positive and negative) resistance effect of each of the significant SSR markers (see Supplementary Table S5 for the effects of all the alleles). In E. grandis, the allele Embra345–225 bp had the maximum positive effect (1.67, 83.3%), whereas EUCeSSR0755-274 bp had the maximum negative effect (−1.70, 39.2%; Table 3). In E. tereticornis, the alleles EUCeSSR683-167 bp and Embra333-250 bp had the maximum positive (1.38, 47.0%) and negative (−1.45, 49.2%) effects, respectively (Table 3). In particular, some of the alleles had the opposite phenotypic effect between the two eucalypt species. For EUCeSSR479 for example, the alleles 204, 231, 213 and 222 bp showed a negative effect in E. grandis (Supplementary Table S5) but a positive effect in E. tereticornis (Supplementary Table S6), in spite of the consistently positive effect of 228, 234, 210 and 225 bp in both species. Similarly, Dillon et al.17 found a flip in the effect of a SNP on pulp yield between discovery and verification populations in the eucalypt species Corymbia citriodora (Hook.) K.D. Hill & L.A.S. Johnson. Such reversals in allelic effect can be a reflection of complex associations in which interactions between the associated allele and other factors are at play17.
This study reveals markers and favourable alleles that have potential in marker-assisted selection for resistance to the gall wasp L. invasa, at least in the important tree genus Eucalyptus. Firstly, the significant markers, especially those validated, could be used to track genomic loci of interest. While the SSRs associated in E. grandis provide candidates for verification purpose in other species, the associations verified in E. tereticornis may suggest robust genomic regions underlying gall wasp resistance. Secondly, the favourable alleles could be used as tags of selection for elite genotypes that have assembled the most desirable alleles over all the associated markers. This is extremely useful for species (e.g. many eucalypts) amenable to mass vegetative propagation as clonal cultivation offers the most effective approach to capture genetic gains71. Thirdly, based on the association results, elite parental combinations could be predicted to generate descendants with improved resistance to L. invasa. In this respect, the alleles robust in both E. grandis and E. tereticornis will have greater utility in a breeding programme.
In conclusion, this study presents genomic loci associated with gall-inducing insect resistance for the first time in plants and makes a valuable contribution to our understanding of the genetic basis underlying plant resistance to gall wasps. Seven SSR markers were associated with resistance to L. invasa in E. grandis, of which four associations were verified in E. tereticornis. These markers plus their favourable alleles can be used for marker-assisted selection for L. invasa resistance in Eucalyptus. Nevertheless, considering the quantitative nature of the insect resistance and the small proportion of genome sampled by the SSR loci, further association work should be undertaken to improve the genome coverage of markers by applying new technologies, such as next-generation sequencing72. In addition, as CG approaches have proven to be advantageous for breeding applications17, genome-wide screening of CGs followed by CG-based association mapping should be conducted for gall wasp resistance.
References
Espírito-Santo, M. M. & Fernandes, G. W. How many species of gall-inducing insects are there on Earth, and where are they? Ann. Entomol. Soc. Am. 100, 95–99 (2007).
Raman, A. Gall induction by hemipteroid insects. J. Plant Interact. 7, 29–44 (2012).
Isaias, R. M. S., Oliveira, D. C., Carneiro, R. G. S. & Kraus, J. E. Developmental anatomy of galls in the Neotropics: arthropods stimuli versus host plant constraints. In Neotropical Insect Galls (eds Fernandes, G. W. & Santos, J. C.). 15–34 (Springer & Business Media Dordrecht, 2014).
Bailey, S., Percy, D. M., Hefer, C. A. & Cronk, Q. C. B. The transcriptional landscape of insect galls: psyllid (Hemiptera) gall formation in Hawaiian Metrosideros polymorpha (Myrtaceae). BMC Genomics 16, 943 (2015).
Oates, C. N., Külheim, C., Myburg, A. A., Slippers, B. & Naidoo, S. The transcriptome and terpene profile of Eucalyptus grandis reveals mechanisms of defence against the insect pest, Leptocybe invasa. Plant Cell Physiol. 56, 1418–1428 (2015).
Granett, J., Walker, M. A., Kocsis, L. & Omer, A. D. Biology and management of grape phylloxeram. Annu. Rev. Entomol. 46, 387–412 (2001).
FAO. Eucalypts for Planting. 1 (Food and Agriculture Organization of the United Nations, 1979).
Midgley, S. J. Making a difference: celebrating success in Asia. Aust. For. 76, 73–75 (2013).
Blanche, K. R. Diversity of insect-induced galls along a temperature−rainfall gradient in the tropical savannah region of the Northern Territory, Australia. Aust. Ecol. 25, 311–318 (2000).
Mendel, Z., Protasov, A., Fisherand, N. & La Salle, J. Taxonomy and biology of Leptocybe invasa gen. & sp. n. (Hymenoptera: Eulophidae), an invasive gall inducer on Eucalyptus. Aust. J. Entomol. 43, 101–113 (2004).
Thu, P. Q., Dell, B. & Burgess, T. I. Susceptibility of 18 eucalypt species to the gall wasp Leptocybe invasa in the nursery and young plantations in Vietnam. Sci. Asia 35, 113–117 (2009).
Nyeko, P., Mutitu, E. K., Otieno, B. O., Ngae, G. N. & Day, R. K. Variations in Leptocybe invasa (Hymenoptera: Eulophidae) population intensity and infestation on eucalyptus germplasms in Uganda and Kenya. Int. J. Pest Manag. 56, 137–144 (2010).
Kim, I.-K., Mendel, Z., Protasov, A., Blumberg, D. & La Salle, J. Taxonomy, biology, and efficacy of two Australian parasitoids of the eucalyptus gall wasp, Leptocybe invasa Fisher & La Salle (Hymenoptera: Eulophidae: Tetrastichinae). Zootaxa 1910, 1–20 (2008).
Goud, K. B., Kumari, N. K., Vastrad, A. S., Bhadragoudar, M. & Kulkarni, H. Screening of eucalyptus genotypes against gall wasp, Leptocybe invasa Fisher and La Salle (Hymenoptera: Eulophidae). Karnataka J. Agric. Sci. 23, 213–214 (2010).
Dittrich-Schröder, G., Wingfield, M. J., Hurley, B. P. & Slippers, B. Diversity in Eucalyptus susceptibility to the gall-forming wasp Leptocybe invasa. Agric. For. Entomol. 14, 419–427 (2012).
Luo, J., Arnold, R., Lu, W. & Lin, Y. Genetic variation in Eucalyptus camaldulensis and E. tereticornis for early growth and susceptibility to the gall wasp Leptocybe invasa in China. Euphytica 196, 397–411 (2014).
Dillon, S. K., Brawner, J. T., Meder, R., Lee, D. J. & Southerton, S. G. Association genetics in Corymbia citriodora subsp. variegate identifies single nucleotide polymorphisms affecting wood growth and cellulosic pulp yield. New Phytol. 195, 596–608 (2012).
Michael, T. P. & VanBuren, R. Progress, challenges and the future of crop genomes. Curr Opin. Plant Biol. 24, 71–81 (2015).
Plomion, C. et al. Forest tree genomics: 10 achievements from the past 10 years and future prospects. Ann. For. Sci. 73, 77–103 (2016).
Heffner, E. L., Sorrells, M. E. & Jannink, J. L. Genomic selection for crop improvement. Crop Sci. 49, 1–12 (2009).
Ingvarsson, P. K. & Street, N. R. Association genetics of complex traits in plants. New Phytol. 189, 909–922 (2011).
Khan, M. A. & Korban, S. S. Association mapping in forest trees and fruit crops. J. Exp. Bot. 63, 4045–4060 (2012).
Krothapalli, K. et al. Forward genetics by genome sequencing reveals that rapid cyanide release deters insect herbivory of Sorghum bicolor. Genetics 195, 309–318 (2013).
Lee, J. S. et al. Detection of novel QTLs for foxglove aphid resistance in soybean. Theor. Appl. Genet. 128, 1481–1488 (2015).
Kloth, K. J., Thoen, M. P. M., Bouwmeester, H. J., Jongsma, M. A. & Dicke, M. Association mapping of plant resistance to insects. Trends Plant Sci. 17, 311–319 (2012).
Remington, D. L. et al. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 98, 11479–11484 (2001).
Wilson, L. M. et al. Dissection of maize kernel composition and starch production by candidate gene association. Plant Cell 16, 2719–2733 (2004).
Harjes, C. E. et al. Natural genetic variation in lycopene epsilon cyclase tapped for maize biofortification. Science 319, 330–333 (2008).
Du, Q., Pan, W., Xu, B., Li, B. & Zhang, D. Polymorphic simple sequence repeat (SSR) loci within cellulose synthase (PtoCesA) genes are associated with growth and wood properties in Populus tomentosa. New Phytol. 193, 763–776 (2013).
Guerra, F. P. et al. Association genetics of chemical wood properties in black poplar (Populus nigra). New Phytol. 197, 162–176 (2013).
Silva-Junior, O. B. & Grattapaglia, D. Genome-wide patterns of recombination, linkage disequilibrium and nucleotide diversity from pooled resequencing and single nucleotide polymorphism genotyping unlock the evolutionary history of Eucalyptus grandis. New Phytol. 208, 830–845 (2015).
Wallace, J. G. et al. Association mapping across numerous traits reveals patterns of functional variation in maize. PLoS Genet. 10, e1004845 (2014).
Lamara, M. et al. Genetic architecture of wood properties based on association analysis and co-expression networks in white spruce. New Phytol. 210, 240–255 (2016).
Zhang, P., Zhong, K., Tong, H., Shahid, M. Q. & Li, J. Association mapping for aluminum tolerance in a core collection of rice landraces. Front. Plant Sci. 7, 1415 (2016).
Singh, N. V. et al. Genetic diversity and association mapping of bacterial blight and other horticulturally important traits with microsatellite markers in pomegranate from India. Mol. Genet. Genomics 290, 1394–1402 (2015).
Motilal, L. A. et al. Association mapping of seed and disease resistance traits in Theobroma cacao L. Planta 244, 1265–1276 (2016).
Zhang, K. et al. Development and identification of SSR markers associated with starch properties and β-carotene content in the storage root of sweet potato (Ipomoea batatas L. Front. Plant Sci. 7, 223 (2016).
Kumar, B. et al. Mapping of yellow mosaic virus (YMV) resistance in soybean (Glycine max L. Merr.) through association mapping approach. Genetica 143, 1–10 (2015).
Edzesi, W. M. et al. Genetic diversity and elite allele mining for grain traits in rice (Oryza sativa L.) by association mapping. Front. Plant Sci. 7, 787 (2016).
Grattapaglia, D. et al. Progress in Myrtaceae genetics and genomics: Eucalyptus as the pivotal genus. Tree Genet. Genomes 8, 463–508 (2012).
Myburg, A. A. et al. The genome of Eucalyptus grandis. Nature 510, 356–362 (2014).
Burgess, I. P. & Bell, J. C. Comparative morphology and allozyme frequencies of Eucalyptus grandis Hill ex Maiden and E. saligna Sm. Aust. For. Res. 13, 133–149 (1983).
Song, Z. et al. Genome scans for divergent selection in natural populations of the widespread hardwood species Eucalyptus grandis (Myrtaceae) using microsatellites. Sci. Rep. 6, 34941 (2016).
Song, Z. et al. Genetic diversity and selective loci in Eucalyptus tereticornis populations. Sci. Silvae Sin. 52(9), 39–47 (2016).
Griffin, A. R. & Cotterill, P. P. Genetic variation in growth of out-crossed, selfed and open-pollinated progenies of Eucalyptus regnans and some implications for breeding strategy. Silvae Genet. 37, 124–131 (1988).
Lynch, M. & Walsh, B. Genetics and Analysis of Quantitative Traits. 807–813 (Sinauer Associates Inc., 1998).
Gan, S. et al. Moderate-density molecular maps of Eucalyptus urophylla S. T. Blake and E. tereticornis Smith genomes based on RAPD markers. Genetica 118, 59–67 (2003).
Li, F. et al. Comparative genomics analyses reveal extensive chromosome colinearity and novel quantitative trait loci in Eucalyptus. PLoS ONE 10, e0145144 (2015).
Li, F. & Gan, S. An optimised protocol for fluorescent-dUTP based SSR genotyping and its application to genetic mapping in Eucalyptus. Silvae Genet. 60, 18–25 (2011).
Dieringer, D. & Schlötterer, C. Microsatellite analyzer (MSA): a platform independent analysis tool for large microsatellite data sets. Mol. Ecol. Notes 3, 167–169 (2003).
Bradbury, P. J. et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 2, 2633–2635 (2007).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Gilbert, K. J. et al. Recommendations for utilizing and reporting population genetic analyses: the reproducibility of genetic clustering using the program STRUCTURE. Mol. Ecol. 21, 4925–4930 (2012).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Hardy, O. J. & Vekemans, X. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2, 618–620 (2002).
Breseghello, F. & Sorrells, M. E. Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172, 1165–1177 (2006).
Wen, Z.-X. et al. Association analysis of agronomic and quality traits with SSR markers in Glycine max and Glycine soja in China: II. Exploration of elite allels. Acta Agron. Sin. 34, 1339–1349 (2008).
Arumugasundaram, S., Ghosh, M., Veerasamy, S. & Ramasamy, Y. Species discrimination, population structure and linkage disequilibrium in Eucalyptus camaldulensis and Eucalyptus tereticornis using SSR markers. PLoS ONE 6, e28252 (2011).
Yan, A. et al. Involvement of C2H2 zinc finger proteins in the regulation of epidermal cell fate determination in Arabidopsis. J. Integr. Plant Biol. 56, 1112–1117 (2014).
Nilsson, I. & Hoffmann, I. Cell cycle regulation by the Cdc25 phosphatase family. In Progress in Cell Cycle Research, Vol. 4 (eds Meijer, L., Jézéquel, A. & Ducommun, B.). 107–114 (Kluwer Academic/Plenum Publishers, 2000).
Sharma, M., Pandey, A. & Pandey, G. K. Role of plant U-BOX (PUB) protein in stress and development. Plant Stress 7(S1), 1–9 (2013).
Mølhøj, M., Pagant, S. & Höfte, H. Towards understanding the role of membrane-bound endo-1,4-glucanases in cellulose biosynthesis. Plant Cell Physiol. 43, 1399–1406 (2002).
Ross, P., Mayer, R. & Benziman, M. Cellulose biosynthesis and function in bacteria. Microbiol. Rev. 55, 35–58 (1991).
Nakhamchik, A. et al. A comprehensive expression analysis of the Arabidopsis proline-rich extensin-like receptor kinase gene family using bioinformatic and experimental approaches. Plant Cell Physiol. 45, 1875–1881 (2004).
Humphrey, T. V. et al. PERK–KIPK–KCBP signalling negatively regulates root growth in Arabidopsis thaliana. J. Exp. Bot. 66, 71–83 (2015).
Schlötterer, C. Hitchhiking mapping – functional genomics from the population genetics perspective. Trends Genet. 19, 32–38 (2003).
Du, Q., Gong, C., Pan, W. & Zhang, D. Development and application of microsatellites in candidate genes related to wood properties in the Chinese white poplar (Populus tomentosa Carr.). DNA Res. 20, 31–44 (2013).
Becerra, J. X. The impact of herbivore–plant coevolution on plant community structure. Proc. Natl. Acad. Sci. USA 104, 7483–7488 (2007).
Brown, G. R. et al. Identification of quantitative trait loci influencing wood property traits in loblolly pine (Pinus taeda L.). III. QTL verification and candidate gene mapping. Genetics 164, 1537–1546 (2003).
Griffin, A. R. Deployment decisions-capturing the benefits of tree improvement with clones and seedlings. In Developing the Eucalypt of the Future (ed. Barros, S.). CD (IUFRO, 2001).
Barabaschi, D. et al. Next generation breeding. Plant Sci. 242, 3–13 (2016).
Acknowledgements
We would like to thank Yonghuai Huang and Yan Lin for assistance with leaf sampling as well as Xiaoli Yu for help in the laboratory. We also thank Simon G Southerton for his critical reading and valuable comments on the manuscript. This work was financially supported by the Department of Science and Technology of Guangxi Zhuang Autonomous Region, China (Guike AA17204065-6), the National Natural Science Foundation of China (31270702) and Guangdong Provincial Department of Science and Technology (2014B020202013).
Author information
Authors and Affiliations
Contributions
S.G. and F.L. conceived and designed the project. M.Z., X.M., H.H., F.L. and S.G. collected the samples of E. grandis. C.Z., Z.S., Q.W. and J.L. collected the samples of E. tereticornis. M.Z., C.Z., Z.S., Q.W., M.L. and H.J. performed molecular labwork and scored the markers. S.G., F.L., M.Z., C.Z. and Z.S. analysed the data and wrote the manuscript with assistance from all other authors. All authors read and approved final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, M., Zhou, C., Song, Z. et al. The first identification of genomic loci in plants associated with resistance to galling insects: a case study in Eucalyptus L'Hér. (Myrtaceae). Sci Rep 8, 2319 (2018). https://doi.org/10.1038/s41598-018-20780-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20780-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
