
Abstract
Colitis, an inflammatory disease of the digestive tract, is increasing in incidence and prevalence. Intestinal inflammation can occur as a consequence of dysfunctions in sphingolipid metabolism. In this study we used ceramide synthase 6 (CerS6) deficient mice, which have a reduced ability to generate long chain C16-ceramide, to investigate the role of this enzyme in dextran sodium salt (DSS)-induced colitis. While CerS6-deficient mice are protected from T cell mediated colitis, in the T cell independent DSS model lack of CerS6 resulted in a more rapid onset of disease symptoms. CerS6-deficient mice maintained low levels of C16-ceramide after DSS treatment, but the inflammatory lipid sphingosine-1-phosphate was significantly increased in colon tissue. In the absence of CerS6, DSS induced more severe pathology in the colon including enhanced neutrophil infiltration. In vivo analysis of myeloperoxidase activity, an enzyme released from neutrophils, was approximately 2.5-fold higher in CerS6-deficient mice compared to wild type. Differences in intestinal permeability did not account for the increase in neutrophils. Our study suggests that lack of CerS6 expression differentially impacts the development of colitis, depending on the model used.
Similar content being viewed by others


Introduction
Inflammatory bowel diseases are emerging as a global problem with increased incidence and prevalence in numerous countries1. Sphingolipid metabolism has been shown to play a role in colitis and the bioactive lipid sphingosine-1-phosphate (S1P) is emerging as a novel therapeutic target2,3,4. S1P is generated by two sphingosine kinases (SK1, SK2) and SK1 has specifically been associated with colitis in the DSS model as SK1-deficient mice are protected and the novel SK1 inhibitor LCL-351 reduces immune responses5,6. S1P is generated from ceramide, which is central to sphingolipid metabolism and varies in acyl chain length ranging from 14–30 (or more) carbons7. This variety in acyl chain length derives from the activity of six ceramide synthases (CerS) with different substrate preferences8. Through the combined activity of multiple CerS, cells are endowed with a specific ceramide profile presumably required for maintenance of sphingolipid homeostasis in any given tissue. Alterations in ceramide profiles have been observed in multiple diseases, suggesting that dysregulation of CerS may contribute to onset or progression of pathologies9,10.
Ceramide synthase 6 (CerS6) is an enzyme that preferentially utilizes 16-carbon fatty acids to generate C16-ceramide. CerS6 mRNA is highly expressed in the intestine and immune system8. CerS6-deficient mice are viable and except for some behavioral abnormalities, do not display any obvious defects under normal conditions11. Different pathologies emerge in disease models however. Lack of CerS6 increases aggressiveness of experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis, diminishes macrophage infiltration and pro-inflammatory gene expression in the diet-induced obesity model, and reduces graft-vs-host disease12,13,14. These opposing consequences of CerS6-deficiency across models underscore the complexity of sphingolipid signaling. Recently, adoptive transfer of CerS6-deficient naïve CD4+ T cells was shown to protect mice from colitis12,15. In this study, we further elucidate the role of CerS6 in DSS-induced colitis, which is independent of T lymphocytes.
Results
Wild type and CerS6-deficient mice develop DSS-induced colitis
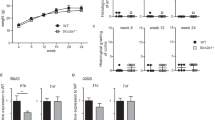
Colitis was induced by adding 3% DSS for 5 days to the drinking water, followed by 3 days of regular water. Body weight and appearance of feces were monitored daily throughout the 8 day experiment. Beginning on day 4, both wild type and CerS6 deficient mice receiving water supplemented with DSS experienced significant weight loss (Fig. 1a). There was no difference in weight loss between wild type and CerS6 deficient mice or between genders. In addition to weight, we monitored the development of diarrhea and assigned a disease activity score. Data analysis using the student t test indicated that CerS6-deficient mice had significantly higher disease activity on days 4 and 5 compared to wild type mice (p = 0.0007 for day 4 and p = 0.0001 for day 5). Further statistical analysis demonstrated that the slopes describing disease activity trends differed between groups. In wild type mice, disease activity score increased over time in a linear manner with slopes of 0.25 and 0.24 for days 1 to 5 and 5 to 8, respectively. In CerS6-deficient mice disease activity index increased more rapidly over the first 5 days (slope 0.54) and then remained relatively constant (slope -0.11) (Fig. 1b). Thus DSS-induced colitis resulted in similar weight loss and disease activity index by day 8 but progressed with different kinetics in wild type and CerS6-deficient mice.

DSS-induced colitis in wild type and CerS6-deficient mice. (a) Body weight and (b) disease activity score based on analysis of feces. Data shown are from 3 independently performed experiments, each with at least n = 4 per group. The untreated group in (a) included both wild type and CerS6-deficient animals. In (b), the difference in slopes prior to day 5 was p = 0.0001 and p = 0.0007 between days 5–8.
Sphingosine-1-phosphate is increased in colons of CerS6-deficient mice
Next, we analyzed sphingolipids in DSS treated mice. MALDI imaging mass spectrometry (IMS) is a powerful tool to analyze the distribution of proteins and other molecules including sphingolipids in tissue sections in situ16,17. CerS6-deficient mice have significantly reduced C16-sphingolipids11,15. This reduced level of C16-sphingolipids was also observed in mice exposed to DSS (Fig. 2). Sphingosine-1-phosphate (S1P), a pro-inflammatory sphingolipid that has been associated with colitis2, was below limits of detection by MALDI IMS probing. Analysis of serum and tissues by LC-MS showed a similar decrease in C16-ceramide (Table 1). Other ceramide species were not significantly altered. Sphingosine did not significantly differ either in circulation or in colon tissue. In untreated wild type or CerS6-deficient mice, serum sphingosine levels are about 25 ng/ml. On day 8 of the DSS protocol, serum sphingosine in wild type mice was 32 ± 8 ng/ml and in CerS6-deficient mice it was 25 ± 7 ng/ml (n = 5). Serum levels of S1P were comparable between wild type and CerS6-deficient mice but a significant increase in S1P was detected in colon tissues of CerS6-deficient mice (Table 1).

MALDI-IMS of colon tissue following induction of DSS colitis. Colon tissue of DSS treated mice was analyzed by MALDI-IMS and signal (area under the curve, AUC) analyzed for C16-ceramide (a) and (b) C16-sphingomyelin. Tissue from wild type and CerS6-deficient mice are indicated by green and red outlines, respectively. This experiment was performed with two sets of female littermates. The white dashed line separates the two sets. *p < 0.05
Neutrophil infiltration is increased in CerS6-deficient mice
Analysis of colon tissue from DSS treated wild type and CerS6-deficient mice indicated that lack of CerS6 was associated with an increased overall pathology score (Fig. 3a). The increased score was primarily driven by a significant increase in abnormal crypts and neutrophil infiltration (Fig. 3b,c). Histopathology of wild type and CerS6-deficient mice is shown in Fig. 4. CerS6-deficient mice exhibited crypt disruption and ectasia with crypt abscesses. Crypt disruption was also observed in wild type mice but to a lesser degree and with only some ectasia and hyperplasia. The lamina propria of CerS6-deficient mice contained numerous mononuclear cells and neutrophils. As in CerS6-deficient mice, neutrophils in wild type animals were most commonly adjacent to ulcerated areas. However, there are fewer areas of ulceration and epithelial disruption in wild type compared to CerS6-deficient mice.

Histological analysis of the colon. (a) Total pathology score, (b) neutrophil infiltration, (c) abnormal crypts. The maximal total pathology score is 17. Neutrophil infiltration and abnormal crypts are sub-score categories of the total score. Data is from 3 independently performed experiments, each with at least n = 4 per group. *p < 0.05

H&E stained colon tissue from DSS treated mice. Images of colon pathology from wild type (a–d) and CerS6-deficient mice (e–h) on day 8 of the experiment. Left panels are 10× and right panels are 20× magnification of the same tissue section. Bars indicate 100 μm. Neutrophils in wild type mice are most commonly found adjacent to ulcerated areas (a,b). Crypt disruption, some ectasia of crypts and some hyperplasia are evident in wild type mice (c,d). Lamina propria of CerS6-deficient mice contain numerous mononuclear cells and neutrophils with most neutrophils are seen in areas of epithelial disruption (e,f). CerS6-deficient mice have evidence of crypt disruption, ectasia with crypt abscesses, and numerous mononuclear cells in the lamina propria (g,h).
To confirm differences in neutrophil infiltration, a subset of mice was analyzed for myeloperoxidase (MPO) activity. Mice were injected with a chemiluminescent in vivo reagent designed to monitor inflammation by detecting myeloperoxidase (MPO) activity of activated phagocytes. Imaging indicated that MPO activity was limited to the intraperitoneal cavity (Fig. 5). MPO activity in CerS6-deficient mice was estimated to be 2.5 times higher than in wild type mice (95% CI: 1.19, 5.28; p = 0.0036).

Bioluminescent imaging of myeloperoxidase activity. A subset of mice was injected with a chemiluminescent substrate that detects MPO activity. Mice were imaged 10 minutes post i.p. injection using a 5 minutes exposure time on days 4, 5, and 6. Cages 1 and 3 were female mice. Cage 2 were male mice. Statistical analysis was performed on photons/second from each image.
Intestinal permeability in CerS6-deficient mice is similar to wild type mice
Pathology and the MPO functional assay suggested that DSS-induced colitis significantly increased the presence of neutrophils in the intestines of CerS6-deficient mice. We therefore investigated the possibility that CerS6-deficient mice have an overall increase in intestinal permeability. Intestinal permeability was assessed by quantifying fecal albumin18,19. Fecal albumin levels did not differ between wild type and CerS6-deficient mice prior to DSS treatment (Fig. 6a). In DSS treated mice, levels of albumin increased substantially but to similar extent in both groups of mice at day 8 (Fig. 6a). Evaluation of fecal albumin between days 3–5 during which disease activity scores are higher in CerS6-deficient mice also failed to reveal any significant differences in intestinal permeability (Fig. 6b).

Gut permeability as a measure of fecal albumin. Mouse fecal albumin was quantified by ELISA in untreated mice (n = 2–3) or at the conclusion of the experiment (n = 5) (a) and on day 3–5 of DSS induced colitis (3 independently performed experiments each with at least n = 4 per group) (b). Data shown are the average ± standard deviation. Differences between wild type and CerS6-deficient mice were not significant.
Discussion
In this study we analyzed the effect of CerS6-deficiency on susceptibility to DSS-induced colitis. Analysis of weight loss suggested there is no difference between wild type and CerS6-deficient mice (Fig. 1a), yet the kinetics of disease activity score indicated a more rapid progression of colitis in CerS6-deficient mice (Fig. 1b). This result was unexpected, since CerS6-deficient mice are protected from development of colitis in the adoptive transfer model12,15. C57BL/6 mice are highly susceptible to DSS colitis, thus weight loss may occur at the maximal rate masking differences in disease progression20. Future experiments using a mild induction of colitis with 1% DSS may reveal differences in weight loss and could also serve as a model to study the role of T cells during the recovery phase21. This is especially relevant as the DSS model, which is classically used as a model of colitis, has recently been recognized as a model that more accurately represents tissue injury.
DSS treated CerS6-deficient mice had reduced levels of C16-sphingolipids and the pro-inflammatory sphingolipid S1P was elevated within colon tissue but not in serum (Table 1). A decrease in C16-sphingolipids is expected in CerS6-deficient mice, although an increase in S1P has not previously been observed at baseline in liver, lung or kidneys15. We therefore hypothesize that increased S1P in colon tissue of DSS treated CerS6-deficient mice is associated with colitis related inflammation. However, future experiments will be required to compare S1P within colon tissue of both untreated and DSS treated mice over the course of disease development.
CerS2 and CerS6 have previously been observed to have opposing effects with regard to apoptotic susceptibility22. Similar opposing effects were observed in the EAE model, in which CerS2 null mice were protected while CerS6 null mice had exacerbated disease14,23. Different phenotypes in the same model system can be expected, since CerS2-deficient mice, which lack the ability to generate very long chain ceramides such as C24-ceramide, have a compensatory increase in C16-ceramide while CerS6-deficient mice have reduced C16-ceramide11,24. Thus it is somewhat surprising that both CerS2- and CerS6-deficient mice are more susceptible to DSS-induced colitis25. However, the mechanisms that lead to increased colitis appear to be quite distinct. In CerS6-deficient mice, circulating levels of sphingosine or S1P were comparable at both baseline or following DSS treatment. In contrast, circulating levels of sphingosine and S1P are significantly increased even prior to DSS treatment in CerS2-deficient mice25. Oertel et al. showed that CerS2-deficiency decreases expression of tight junction proteins, which increases gut permeability25. This increase in intestinal permeability leads to elevated immune activation even in the absence of DSS treatment25. In CerS6-deficient mice, we did not detect a difference in intestinal permeability prior to DSS exposure and although fecal albumin levels increased with exposure to DSS there was no significant difference between wild type and CerS6-deficient mice (Fig. 6). Thus increased gut permeability could not account for the more rapid increase in disease activity score in CerS6-deficient mice prior to day 5.
Chemical disruption of the gut mucosa with DSS induces inflammation in the absence of B or T cells, although T cells have been shown to play a role in the recovery phase following induction of mild colitis26,27. Administration of 3% DSS results in severe colitis with acute inflammation of the colon involving infiltration of granulocytes and macrophages in the mucosa28. Macrophages may have played a more prominent role in one wild type animal, which had an average pathology score but no evidence of neutrophil infiltration. In general however, the early influx of neutrophils in the colon has been shown to correlate with tissue destruction on day 821. Histological analysis of colons on day 8 in our study indicated that CerS6-deficient mice had an overall higher pathology score associated with significantly more abnormal crypts and enhanced neutrophil infiltration (Figs 3 and 4). Significantly increased neutrophil MPO activity in CerS6-deficient mice was detected as early as day 4 (Fig. 5). The underlying reason for the enhanced colitis and/or neutrophil infiltration in CerS6-deficient mice will require further analysis. In vitro experiments with CerS6-deficient neutrophils have shown that stimulation with IFNγ/TNFα increases nitric oxide, CD11b expression, and adhesion while stimulation with G-CSF increases migration through upregulation of CXCR2 expression11,14. The increase in CD11b and CXCR2 expression in CerS6-deficient neutrophils was also observed in an in vivo model of multiple sclerosis (EAE model)14. In this model, CerS6-deficient mice experienced exacerbated disease that was associated with increased neutrophil infiltration into the central nervous system14. Cytokines and chemokines (CCL2, CCL5, CXCL2, IL-17, Il-6) as well as iNOS were also significantly increased upon EAE induction in CerS6-deficient mice14. We speculate that neutrophils in DSS treated CerS6-deficient mice may have a similar increase in CD11b and CXCR2, which would be indicative of enhanced activation and migration, and offer a potential explanation for the more rapid onset of disease activity during days 1 and 5 (Fig. 1b).
In conclusion, we have shown that CerS6-deficient mice are more susceptible to DSS-induced colitis, which contrasts with the protective effect that lack of CerS6 has in adoptive transfer models of colitis. Studies with CerS6-deficient mice suggest that this protein has different functions in distinct immune cell subsets, which will complicate efforts to develop specific inhibitors. Generation of tissue-specific CerS knockout mice is expected to further elucidate the roles of CerS family members.
Methods
Animals and DSS induced colitis
All animal experiments were performed with approval by the Institutional Animal Care and Use Committee at the Medical University of South Carolina to ensure that ethical regulatory and policy mandates governing the use of animals in research are met (Animal Welfare Assurance #A3428-01). All methods were performed in accordance with the relevant guidelines and regulations. CerS6-deficient mice (C57BL6) were generated by the Texas Institute of Genomic Medicine and obtained from Dr. Besim Ogretmen. Mice were maintained through heterozygous breeding. Genomic tail DNA was used to genotype offspring by PCR. Primers were as follows: 5′ TTCGGTTAAGAATGGCCTTG3′; 5′CACACCCATATGGAACTCTTACA-3′; and 5′-CCAATAAACCCTCTTGCAGTTGC-3′. Expected PCR products are 460 bp for wild type, 295 bp for CerS6 knockout, or both for heterozygous animals. The C57BL/6 strain is highly susceptible to DSS colitis with no significant differences in the frequency of ulceration between sexes20. To induce colitis, 3% DSS w/v (MP Biomedicals, cat no. 160110) was added to the drinking water for 5 days with a water change on day 3. Mice were returned to regular drinking water on day 5. During the experiment weight was monitored daily and feces collected. The percentage of weight loss was calculated by dividing body weight on the specified day in accordance with body weight on day 0 (baseline). Disease activity scores were assigned as follows: 0 = normal stool; 1 = soft stool; 2 = very soft stool with blood; 2.5 = watery stool but no rectal bleeding; 3 = watery stool with rectal bleeding. On day 8 mice were humanely euthanized and colon tissue prepared for histological analysis, LC-MS or MALDI-IMS. Serum was collected for LC-MS.
Bioluminescent Imaging
In vivo imaging studies fur was removed with depilatory cream for better visualization of the signal. The XenoLight RediJect Inflammation Probe (Perkin-Elmer, cat no. 760536) was injected intraperitoneal (i.p.) at a concentration of 200 mg/kg. Mice were imaged using the IVIS200 system at 10 minutes post i.p. injection using an exposure time of 5 minutes. MPO activity (photons/second) was quantified using Living Image software.
Histology analysis
Colons were removed, cleaned, and fixed in formalin. Sections were placed in cassettes and cut longitudinally for histological analysis. Tissues were analyzed for lamina propria inflammation (0–3), goblet cell loss (0–2), abnormal crypts (0–3) and crypt abscesses (0–1), mucosal erosion and ulceration (0–1), submucosal spread to transmural involvement (0–3) and the number of neutrophils counted at x40 magnification (0–4) for a maximal score of 1729.
Sphingolipid analysis by LC-MS and MALDI-IMS
Ceramide species (C14, C16, C18, C18:1, C20, C20:1, C20:4, C22, C22:1, C24, C24:1, C26 and C26:1 were quantified by LC-MS as previously described30. For MALDI-IMS, colons were collected, feces removed, and tissue wrapped into aluminum foil and flash frozen in liquid nitrogen vapor and stored at −80 °C until analysis of lipids by the MUSC Proteomics Center. Tissues were processed by embedding them into chilled 2% carboxymethylcellulose and flash frozen using dry ice powder. Following overnight storage at −80 °C, cryosections were cut at 10 µm and thaw mounted onto ethanol-cleaned indium tin oxide coated microscope slides. Tissues on slides were coated with a chemical matrix (25 mg/mL 2, 5-dihydroxybenzoic acid with 3 mM KCl and 70% methanol in 0.1% trifluoroacetic acid). Matrix was sprayed onto tissue using an automated sprayer (TM-Sprayer, HTXImaging) with 10 passes at 60 °C, 3 mm offset, 1200 velocity, 10 psi and a 40 mm distance nozzle tip to slide surface. Data were acquired in positive ion mode using a Fourier Transform Ion Cyclotron Resonance (FT-ICR) mass spectrometer (7 Tesla solariX, Bruker Daltonics) equipped with a matrix-assisted laser desorption ionization (MALDI) source. The SmartBeam II laser settings used a total of 500 shots per pixel operated using the smartwalk feature set to 25 µm with one scan per pixel. Stepsize between sampling was 100 µm. Time domain was set to 1 M word with a mass range of 200–1,600 m/z, resulting in a 1.2059 transient with a calculated resolving power of 160,000 at m/z 400. Ion accumulation time was 0.1 second. Data were processed and visualized using SCiLS Lab Imaging Software (Bruker Daltonics, version 2017a. Databases of lipid classes were made from LipidMaps by searching “ceramides” (42 species), “sphingomyelins” (19 species), lyso-glycerophosphocholines (6 species), and lyso-glycerophosphoethanolamines (27 species). Accurate masses of lipids with adducts (M + H, M + Na, M + K, M-H2O + H) were calculated using the software IsotopePattern (Bruker Daltonics).
Albumin ELISA
Feces were dissolved in sample dilution buffer (50 mM Tris, 0.14 M NaCl, 0.05% Tween20, pH 8.0) to a concentration of 100 mg/ml. Albumin levels were quantified using the mouse albumin ELISA kit (Bethyl Labs) according manufacturer instructions and results quantified using a FluoStar Optima plate reader.
Statistical Analysis
Two-sample t-tests were used to compare means between wild type and CerS6-deficient groups. Statistical significance was defined by a two-sided alpha level of 0.05. Disease activity score was analyzed with mixed effects linear regression to account for repeated measures over time. A model was fit, which allowed for each group to have a slope between day 1 and day 5, and another slope between day 5 and day 8 (i.e. a spline model with a knot at 5 days and interactions between time effect and group (WT vs. CerS6 KO). Wald tests were used to compare slopes between groups for each time period. For analysis of bioluminescent imaging, a 3-factor repeated measures Anova was used to evaluate difference between inflammation in wild type and CerS6-deficient mice, accounting for trends over time and cage effects. Due to skewness, a log transform of MPO activity was used as the outcome variable.
References
Ye, Y., Pang, Z., Chen, W., Ju, S. & Zhou, C. The epidemiology and risk factors of inflammatory bowel disease. International journal of clinical and experimental medicine 8, 22529–22542 (2015).
Degagne, E. & Saba, J. D. S1pping fire: Sphingosine-1-phosphate signaling as an emerging target in inflammatory bowel disease and colitis-associated cancer. Clinical and experimental gastroenterology 7, 205–214, https://doi.org/10.2147/CEG.S43453 (2014).
Danese, S., Furfaro, F. & Vetrano, S. Targeting S1P in Inflammatory bowel disease: new avenues for modulating intestinal leukocyte migration. Journal of Crohn’s & colitis. https://doi.org/10.1093/ecco-jcc/jjx107 (2017).
Peyrin-Biroulet, L., Christopher, R., Behan, D. & Lassen, C. Modulation of sphingosine-1-phosphate in inflammatory bowel disease. Autoimmunity reviews 16, 495–503, https://doi.org/10.1016/j.autrev.2017.03.007 (2017).
Pulkoski-Gross, M. J. et al. Novel sphingosine kinase-1 inhibitor, LCL351, reduces immune responses in murine DSS-induced colitis. Prostaglandins & other lipid mediators 130, 47–56, https://doi.org/10.1016/j.prostaglandins.2017.03.006 (2017).
Snider, A. J. et al. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 23, 143–152, https://doi.org/10.1096/fj.08-118109 (2009).
Mullen, T. D., Hannun, Y. A. & Obeid, L. M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem J 441, 789–802, https://doi.org/10.1042/BJ20111626 (2012).
Levy, M. & Futerman, A. H. Mammalian ceramide synthases. IUBMB life 62, 347–356, https://doi.org/10.1002/iub.319 (2010).
Grosch, S., Schiffmann, S. & Geisslinger, G. Chain length-specific properties of ceramides. Progress in lipid research 51, 50–62, https://doi.org/10.1016/j.plipres.2011.11.001 (2012).
Park, W. J. & Park, J. W. The effect of altered sphingolipid acyl chain length on various disease models. Biological chemistry 396, 693–705, https://doi.org/10.1515/hsz-2014-0310 (2015).
Ebel, P. et al. Inactivation of ceramide synthase 6 in mice results in an altered sphingolipid metabolism and behavioral abnormalities. The Journal of biological chemistry 288, 21433–21447, https://doi.org/10.1074/jbc.M113.479907 (2013).
Sofi, M. H. et al. Ceramide synthesis regulates T cell activity and GVHD development. JCI insight 2, https://doi.org/10.1172/jci.insight.91701 (2017).
Turpin, S. M. et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell metabolism 20, 678–686, https://doi.org/10.1016/j.cmet.2014.08.002 (2014).
Eberle, M. et al. Exacerbation of experimental autoimmune encephalomyelitis in ceramide synthase 6 knockout mice is associated with enhanced activation/migration of neutrophils. Immunology and cell biology 93, 825–836, https://doi.org/10.1038/icb.2015.47 (2015).
Scheffel, M. J. et al. Adoptive Transfer of Ceramide Synthase 6 Deficient Splenocytes Reduces the Development of Colitis. Sci Rep 7, 15552, https://doi.org/10.1038/s41598-017-15791-x (2017).
Angel, P. M. & Caprioli, R. M. Matrix-assisted laser desorption ionization imaging mass spectrometry: in situ molecular mapping. Biochemistry 52, 3818–3828, https://doi.org/10.1021/bi301519p (2013).
Jones, E. E. et al. On-tissue localization of ceramides and other sphingolipids by MALDI mass spectrometry imaging. Analytical chemistry 86, 8303–8311, https://doi.org/10.1021/ac501937d (2014).
Hartmann, P. et al. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology 58, 108–119, https://doi.org/10.1002/hep.26321 (2013).
Wang, L. et al. Methods to determine intestinal permeability and bacterial translocation during liver disease. Journal of immunological methods 421, 44–53, https://doi.org/10.1016/j.jim.2014.12.015 (2015).
Mahler, M. et al. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. The American journal of physiology 274, G544–551 (1998).
Hall, L. J. et al. Induction and activation of adaptive immune populations during acute and chronic phases of a murine model of experimental colitis. Digestive diseases and sciences 56, 79–89, https://doi.org/10.1007/s10620-010-1240-3 (2011).
Mesicek, J. et al. Ceramide synthases 2, 5, and 6 confer distinct roles in radiation-induced apoptosis in HeLa cells. Cellular signalling 22, 1300–1307, https://doi.org/10.1016/j.cellsig.2010.04.006 (2010).
Barthelmes, J. et al. Lack of ceramide synthase 2 suppresses the development of experimental autoimmune encephalomyelitis by impairing the migratory capacity of neutrophils. Brain, behavior, and immunity 46, 280–292, https://doi.org/10.1016/j.bbi.2015.02.010 (2015).
Laviad, E. L. et al. Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. The Journal of biological chemistry 283, 5677–5684, https://doi.org/10.1074/jbc.M707386200 (2008).
Oertel, S. et al. Ceramide synthase 2 deficiency aggravates AOM-DSS-induced colitis in mice: role of colon barrier integrity. Cellular and molecular life sciences: CMLS., https://doi.org/10.1007/s00018-017-2518-9 (2017).
Tlaskalova-Hogenova, H. et al. Involvement of innate immunity in the development of inflammatory and autoimmune diseases. Annals of the New York Academy of Sciences 1051, 787–798, https://doi.org/10.1196/annals.1361.122 (2005).
Wang, L., Jiang, X., Liu, X., Qian, T. & Chu, Y. Local immune compartments are related to the severity of dextran sodium sulphate induced colitis. Bioscience trends 8, 242–247 (2014).
Dieleman, L. A. et al. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 107, 1643–1652 (1994).
Ostanin, D. V. et al. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. American journal of physiology. Gastrointestinal and liver physiology 296, G135–146, https://doi.org/10.1152/ajpgi.90462.2008 (2009).
Tirodkar, T. S. et al. Expression of Ceramide Synthase 6 Transcriptionally Activates Acid Ceramidase in a c-Jun N-terminal Kinase (JNK)-dependent Manner. The Journal of biological chemistry 290, 13157–13167, https://doi.org/10.1074/jbc.M114.631325 (2015).
Acknowledgements
The authors would like to thank the Hollings Cancer Center shared resources facilities for assistance with lipidomics analysis and biostatistics. Shared Resources of the Hollings Cancer Center, Medical University of South Carolina were in part supported by P30 CA138313. We would also like to thank Margaret Romano at the Pathology Core Facility for preparation of fixed tissue for histological analysis. The research presented in this article was supported in part by pilot research funding from the COBRE in Lipidomics and Pathobiology grant (5P30GM103339) and a Program Project grant (P01CA203628) at the Medical University of South Carolina.
Author information
Authors and Affiliations
Contributions
C.V.J. was responsible for experimental design, manuscript preparation, preparation of tissues for MALDI-IMS and the albumin ELISA. P.L. performed all animal experiments, collected serum and prepared samples for LC-MS. K.H. was responsible for analysis of histology. B.O. developed the CerS6-deficient mice. EGM performed statistical analysis. R.D. and P.A. performed MALDI-IMS. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Helke, K., Angel, P., Lu, P. et al. Ceramide Synthase 6 Deficiency Enhances Inflammation in the DSS model of Colitis. Sci Rep 8, 1627 (2018). https://doi.org/10.1038/s41598-018-20102-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20102-z
This article is cited by
-
Functional roles of sphingolipids in immunity and their implication in disease
Experimental & Molecular Medicine (2023)
-
Untargeted metabolomics identifies potential serum biomarkers associated with Crohn’s disease
Clinical and Experimental Medicine (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
