
Abstract
With fossil representatives from the Silurian capable of respiring atmospheric oxygen, millipedes are among the oldest terrestrial animals, and likely the first to acquire diverse and complex chemical defenses against predators. Exploring the origin of complex adaptive traits is critical for understanding the evolution of Earth’s biological complexity, and chemical defense evolution serves as an ideal study system. The classic explanation for the evolution of complexity is by gradual increase from simple to complex, passing through intermediate “stepping stone” states. Here we present the first phylogenetic-based study of the evolution of complex chemical defenses in millipedes by generating the largest genomic-based phylogenetic dataset ever assembled for the group. Our phylogenomic results demonstrate that chemical complexity shows a clear pattern of escalation through time. New pathways are added in a stepwise pattern, leading to greater chemical complexity, independently in a number of derived lineages. This complexity gradually increased through time, leading to the advent of three distantly related chemically complex evolutionary lineages, each uniquely characteristic of each of the respective millipede groups.
Similar content being viewed by others


Introduction
One of the most significant events in the evolution of early life on planet Earth took place in the middle Silurian —roughly 423 million years ago— with the origin of the first land animals1,2, triggering the greatest documented expansion of species-level diversity3. Because arthropods are the oldest land animals, and the most species-rich group ever to inhabit the planet, the evolution of chemical defenses largely employed now against predators, likely played a major role in their early diversification. Traits shaped by co-evolutionary interactions between species, such as defensive mechanisms, are often complex morphological/physiological systems; how these systems originated through the action of mutation, drift and natural selection is a central question in evolutionary biology4. Despite the complexity and importance of arthropod chemical defense systems, most studies have focused on understanding the signaling of defense against predators5,6 rather than the evolution of chemical defenses themselves7, therefore their origin and mode of evolution is yet fully understood. Here we investigate the evolution of complex chemical defense systems in the arthropod class Diplopoda (millipedes) using an interdisclipinary approach that employs analytical chemistry and modern genomics-based approaches to generate an evolutionary, phylogenetic framework to investigate chemical defense evolution across earth’s oldest known terrestrial animal group.
Simply stated, the central paradigm for the evolution of complexity is that natural selection drives a gradual elaboration towards multi-part complex traits, with intermediate steps between simple and complex structures8,9,10. Such a stepwise progression towards complexity is not widely accepted because of shortcomings associated with the assumption that evolution proceeds from simple to complex in light of the many documented cases where evolution results in loss or reduced functionality of structures10. Nonetheless, step-wise evolution from simple to complex systems has been observed at the anatomical, behavioral, molecular and metabolic level11,12,13,14. At the metabolic level, for example, the addition of reaction pathways has been observed to provide a stepping-stone model-like approach toward complex metabolic features in the face of nascent evolutionary pressures14.
Here we study the chemicals and pathways involved in millipede defense systems (Diplopoda), their phylogenetic comparative distributions, and hypothesized mode of evolution. Millipedes (class Diplopoda, Fig. 1) are the oldest known fully terrestrial animal group1,15 and are the most diverse class within the Myriapoda with approximately 12,000 known species worldwide and between 3,000 to 80,000 remaining to be described16. Furthermore, millipedes are ubiquitous in forest ecosystems, performing an important role as soil detritivores17,18. All but five millipede orders have repugnatorial glands that secrete chemical defenses when disturbed by predators19. These chemicals belong to at least eight molecule types (i.e., 1,4-benzoquinones, phenols, hydrogen cyanide, quinazolinones, and alkaloids, see Fig. 2)20. The most complex chemical system within millipedes—in terms of diversity of chemical structure and associated anatomy—is found in Juliformia (see Fig. 1) with the production of benzoquinones (Fig. 2, green blue clade). Juliforms produce a variety of other compounds not involved in the benzoquinone pathway like fatty acid esters and aliphatic compounds20. This system ranges from families that produce two chemical molecules (e.g. Nemasomatidae, Parajulidae, Cambalidae, see SI Appendix, Table S1.5) to families that can produce over 1620,21. As new molecule types are discovered and attribute greater chemical complexity to individual millipede families, there is clearly an observed pattern of a positive directional increase within chemical pathways through evolutionary time (Fig. 2). For example, phenol has been found to be the most widely shared chemical type, being produced by some species in six of the seven eugnathan orders in Callipodida, Spirobolida, Spirostreptida, Julida, Stemmiulida and Polydesmida (Figs 1 and 2). Studies on the evolution of phenol secretions in other arthropod groups have recently produced surprising results suggesting a single origin for its production, ancestral to quinones22,23, which indicates that phenols are liklely to be quinone precursors, and the first step in the benzoquinone pathway (Fig. 2). Studies focusing on patterns of chemical production in millipedes have led to the same hypothesis20,24, but in the absence of a well-sampled phylogenetic framework, proper ancestral character mapping methods and models of character evolution.

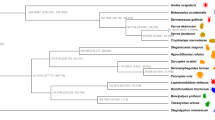
Maximum likelihood tree with branch lengths obtained from analysis of matrix M1. Tree is rooted with Symphylella vulgaris. Bootstrap support (BS) values for all analyses are summarized. BS values not indicated are 100%. Boxplots indicate bootstrap value ranges for each node across all analyses (A–J). Black boxes represent 90–99% BS, dark grey boxes 70–89% BS, light grey boxes BS less than 69% BS, and white boxes where the node whas not recovered for the analysis. (A) ExaML optimal ML tree for matrix M1. (B) ExaML optimal ML tree for matrix M2. (C) ExaML optimal ML tree for matrix M3. (D) ExaML optimal ML tree for matrix M4. (E) ASTRAL greedy consensus tree for matrix M1. (F) ASTRAL greedy consensus tree for matrix M2. (G) ASTRAL greedy consensus tree for matrix M3. (H) ASTRAL greedy consensus tree for matrix M4. (I) ExaML optimal ML tree for matrix M5. (J) ExaML optimal ML tree for matrix M6.

A chronogram for millipedes showing ancestral reconstruction for chemical complexity. (A) Benzoquinone chemical evolution and chemical pathway. Molecular structures on nodes of represent phenol origin and ancestral hydroquinone. Chemical evolution mapped as a continuous character. Colors on phylogeny branches represent number of chemicals produced (A) and correspond to colors encircling molecular structures in the chemical pathway (B). (B) Hypothetical chemical pathway representing the diversity of benzoquinone defense from their corresponding hydroquinone (modified from79). (C) Ancestral chemical ML reconstruction summarized from Figures S1.3–S1.5. Chemical with highest probability (>75%) is shown on the first node reconstructed. [1] Phenol, [2] Hydroquinone, [3] 1,4-Benzoquinone, [4] 2-Methoxyhydroquinone, [5] 2-Methoxy-1,4-benzoquinone, [6] 2-Methoxy-3-hydroxy-1,4-benzoquinone, [7] 2,3-Dimethoxyhydroquinone, [8] 2,3-Dimethoxy-1,4-benzoquinone, [9] 2-Methylhydroquinone, [10] 2-Methyl-1,4-benzoquinone, [11] 2-Methyl-3-hydroxy-1,4-benzoquinone, [12] 2-Methoxy-3-methylhydroquinone, [13] 2-Methoxy-3-methyl-1,4-benzoquinone, [14] 2-Methoxy-3-methyl-5-hydroxy-1,4-benzoquinone, [15] 2-Methoxy-3,6-dimethylhydroquinone, [16] 2-Methoxy-3,6-dimethyl-1,4-benzoquinone, [17] 2,3-dimethoxy-5-methylhydroquinone, [18] 2,3-dimethoxy-5-methyl-1,4-benzoquinone, [19] 2-Ethyl-1,4-benzoquinone, [20] Naphthoquinone, [21] Benzoil cyanide, [22] alpha-terpinene, [23] Polyzonimine.
To determine if millipede generated phenols are ancestral to benzoquinones and to accurately model the sequence of events leading to the evolution of chemical systems in millipedes, a robust phylogeny and a reliable timescale of diversification are needed. However, the age and relationships between the main millipede lineages remain at this time controversial. The fossil record of Paleozoic millipedes is sparse1,25 and suggests that the origin of crown-group millipedes (i.e., most recent common ancestor of all living millipedes) is dated at least at 423 Ma1. Molecular divergence dating studies estimate an older origin for millipedes15,26,27,28. Moreover, various attempts have been made at elucidating the phylogenetic relationships among the orders15,19,29,30. The ordinal relationships and monophyly of higher taxa are still debated because reconstructions using morphological and molecular data have low support or are based on incomplete taxon sampling. Molecular phylogenetic analyses have generated some surprising results, including the paraphyly of Nematophora, and the placement of Stemmiulida as sister to Polydesmida15,27. These past results have key implications for understanding chemical evolution, suggesting that, because of their near-universality in Eugnatha, phenolic compounds are the oldest defense chemicals produced by eugnathan millipedes. Consequently, the conflict between morphological and traditional (Sanger-based) molecular data provides little in the way of reliable answers to some of these long-established evolutionary questions.
To address these issues, we obtained transcriptomic data for 44 millipede taxa from 14 orders to reconstruct the most complete phylogenomic hypothesis for millipedes ever to date. From the literature and newly analyzed data (first reported here), we then determined the chemical composition for six millipede species (Table S1.2) and added information of published chemical records from Shear20 for another 155 species from all but one of the chemically defended orders. For Juliformia we put together data for a total of 35 species, including six newly analyzed. These data indicate the highest diversity of chemical defense molecules occurs in the benzoquinone pathway, produced by the superorder Juliformia. One family from each of the three orders in this group produces more than 15 varieties of quinones (Fig. 2).
Based on a newly derived phylogenetic framework and a synthesis of all the chemical data available to us at this time, the main goal of our study is to test the hypothesis of step-wise evolution from simple to complex8,9,10 in arthropod chemical defense systems using a phylogenetic comparative approach by asking (i) did benzoquinone and cyanogenic production evolve after the ability to produce phenols in eugnathan millipedes and (ii) were the benzoquinone defense systems produced in a step-wise fashion from simple (few molecule types) to complex (many molecule types)?
Results and Discussion
Phylogenetic relationships among main millipede lineages
Although some progress has been made towards inferring phylogenetic relationships among millipede orders15,19,27,28,30, ordinal relationships and monophyly of major clades remain largely unresolved. The most widely supported hypothesis is that Penicillata (bristly millipedes) diverged from Chilognatha early in the history of the group. This hypothesis was traditionally based on the presence of “primitive” characters in Penicillata29,31,32,33,34,35 and has phylogenetic support from both morphological and molecular data27,29. Within Chilognatha, the monophyly of Pentazonia (Glomerida, Sphaerotheriida, Glomeridesmida) has been largely supported, but the monophyly of the Oniscomorpha (Glomerida, Sphaerotheriida), a clade based on the ability to form a sphere and the presence of an enlarged second tergite and anal shield29,36, remains controversial because morphological data support the grouping29,31, whereas molecular and combined datasets render it paraphyletic19,27. These results suggest the ability to roll into a sphere has been secondarily lost in the Glomeridesmida. Volvation is documented in four (Sphaerotheriida, Glomerida, Polyzoniida, Polydesmida) of the 16 millipede orders37 and has evolved repeatedly in many soil-dwelling arthropods. The large clade Helminthomorpha has been proposed based on the presence of gonopods on the seventh body ring in males29 and by the serial lateral repugnatory glands38,39 the monophyly of which has been supported by all phylogenetic analyses to date (15, 19, 27–29). The monophyly of the Colobognatha and its sister-group relationship to the Eugnatha has variously been disputed (as summarized by Hoffman36, but both have been supported by all phylogenetic analyses15,27,29). A placement of the Colobognatha as a tentative sister to the Polydesmida was considered spurious by the authors (19), suggesting further data collection being required. Another result that affects the inference of phenol evolution is the putative monophyly of Eugnatha –supported in all analyses except a single combined morphological/molecular dataset19. All Eugnatha, except for the order Chordeumatida, have families that produce phenols40. This suggests a single origin before benzoquinone and cyanogenic production. One of the most controversial groupings in previous classifications, the Nematophora29,31, was established on the basis of the presence of pre-anal spinnerets41, uniting the orders Callipodida, Chordeumatida and Stemmiulida. The non-monophyly of the Nematophora had been recovered in a total evidence analysis19 and recent phylogenomics studies15,27. The monophyly of the recently proposed “eighth gonopod clade” (ref.42, postulated to contain the orders Callipodida, Chordeumatida, Polydesmida and Stemmiulida) is not confirmed by our current analysis (Fig. 1). The spinnerets of Stemmiulida are morphologically distinct from those of Callipodida and Chordeumatida, but those of Polydesmida are virtually identical to Callipodidan spinnerets, suggesting that the form of the spinnerets in Stemmiulida is an autapomorphy, and that spinnerets have been lost in Juliformia43. It is uncertain whether the modified setae and the epiproct of the Stemmiulida are actually functioning as silk-producing organs43.
To produce a well-sampled phylogeny for the Diplopoda we generated transcriptomes and identified orthologs for 44 taxa across 26 families and 14 orders (average de novo assembled transcripts per species 49,523; average length 773), and combined these with orthologs from 15 published transcriptomes from ingroup and outgroup taxa (SI Appendix, Table S1.1). Our concatenated analyses of matrices M1-M4 largely recover lineages supported by earlier phylogenetic studies29,30. Penicillata is placed as sister to all remaining millipedes, branching off the earliest among the orders. Morphological characters supporting this early branching are the lack of gonopods32, uncalcified cuticle, indirect sperm transfer, well-developed body musculature33, serrated bristles for defense, and hemianamorphosis34 in Penicillata. As in most previous analyses the monophyly of the Chilognatha is recovered, and supported by several morphological characters38,39. Whereas Pentazonia is monophyletic, the monophyly of the Oniscomorpha (Glomerida, Sphaerotheriida) is rejected in all of our analyses, supporting the loss of the ability to roll into a sphere in Glomeridesmida. Helminthomorpha is monophyletic, supported morphologically by the presence of serial lateral pairs of defense glands (lost in Chordeumatida and Siphoniulida) tracheal characters38,39, and the adult being composed of at least 21 body rings29 although many polydesmids comprise 20 segments. Within Helminthomorpha, a monophyletic Colobognatha is sister to a monophyletic Eugnatha. Colobognatha is characterized by simple leg-like gonopods —as well as a number of autapomorphies proposed by Enghoff29: eight legs anterior to the male gonopods, gnathochilarial palps absent, first larval stadium with four pairs of legs, eggs protected by adults by curling around them, defense glands elongate-subtubular. It should be noted that morphological variability within millipedes has not been thoroughly examined across a wide spectrum of taxa19. Within Colobognatha the placement of Siphonophorida had only been established by a combined dataset19, where it was sister to Siphonocryptida + Platydesmida. We present here the first transcriptome data for this order and support its sister relationship to Polyzoniida + Platydesmida (Siphonocryptida is not included in our analysis). Eugnatha is supported by the results in each of our analyses, and is characterized morphologically by the pleurites fused to tergites, subspherical defense glands29, seven pairs of legs in front of the gonopods and an ontogenetic development strikingly different from the ontogeny of the colobognathan gonopods, suggesting non-homology between the colobognathan and eugnathan gonopods30. Within Eugnatha, a clade formed by Callipodida sister to Chordeumatida is sister to all remaining eugnaths. This sister relationship had been proposed by all earlier phylogenetic reconstructions (19, 28, 29) and long established morphologically by the special structure of the Tömösváry organ34. The monophyly of remaining eugnaths, however, has low support in ASTRAL analyses with high amounts of missing data, and in the most phylogenetic informative dataset (M6). This is not surprising given the sensitivity of ASTRAL to non-random missing data44 and the selection of high informativeness to certain time-frames, which could affect informativeness at other points in the phylogeny (see Materials and Methods). Juliformia is herein widely supported as monophyletic and Nematophora is polyphyletic. Juliformia has been established based on the sternites fused to pleurotergites, second larval stadium with seven pairs of legs, collum enlarged, defensive secretions with benzoquinones, and spermatozoa with pseudoperforatorium29. Although the monophyly of Juliformia has never been controversial, the relationships between its three orders had shown divergent results through the years, suggesting all possible combinations of relationships19,31. Our results support the sister relationship between Spirostreptida and Julida, albeit with lower support values for the supermatrix and informativeness analyses. Within Spirostreptida, the position of Cambaloidea had been controversial30. All our analyses include this suborder within Spirostreptida. Enghoff et al.34 suggested the validity of the sister relationship Polydesmida + Juliformia, and referred to it as the “ring forming millipedes”. Our topology infers Juliformia as sister to a clade formed by Stemmiulida and Polydesmida, which is supported by concatenated and ASTRAL analyses with low missing data. Again, this could be explained by the impact of missing data on the accuracy of ASTRAL reconstructions44. The lack of complete body rings in Stemmiulida in light of these relationships suggests a possible reversal of the formation of complete body rings in this order, as well as another reversal in a family of the order Julida, the Nemasomatidae45. Another –less parsimonious– option could be two separate, independent origins for ring formation in Polydesmida and Julimorpha (Fig. 1). Orders not included in this analysis are Siphoniulida and Siphonocryptida. The latter has only been included in a combined dataset and proposed as sister to Platydesmida19. The relationships of Siphoniulida are still obscure. Enghoff29 proposed that it should be considered a subordinate taxon within a juliformian or colobognath order. Despite the discovery of new specimens of this enigmatic group, phylogenetic analyses produced equivocal results, and therefore it is still placed as Helminthomorpha incertae sedis30. Divergence time estimation results suggest an origin for Diplopoda ca. 467 Ma (95% HPD: 424–531). This age is much earlier than suggested by the fossil record, but the youngest bound of the 95% HPD coincides with the oldest diplopod fossil recorded1.
Chemical evolution
To estimate divergence times onto the millipede phylogeny we analyzed a reduced dataset of 38 partitions using a relaxed molecular clock and eight calibration points based on fossil data (SI Appendix, Table S1.4). We also produced a chronogram for the complete phylogeny using dates produced by the aforementioned analysis in a Penalized Likelihood framework46,47. Chemical composition was determined from published records and chemical analyses (SI Appendix, Table S1.2). Chemicals grouped into quinazolinone alkaloids, heterocyclic nitrogen-containing compounds, terpenes, benzoquinones-hydroquinones and phenol, and cyanogenics were mapped as separate characters (Appendix S1, Figures S1.2–S1.5). Phenol and benzoquinone production were mapped as a single discrete character to determine if phenol production was the ancestral condition (Appendix S1, Figure S1.2). Benzoquinone complexity was mapped onto the phylogeny as a continuous character to identify the directionality of chemical evolution (Fig. 2A,B). Our results indicate that the origin of phenol production occurred once during the evolution of millipedes at 315 Ma when Callipodida and Chordeumatida diverged from remaining eugnaths or twice with independent origin in Callipodida and the MRCA of Juliformia, Stemmiulida and Polydesmida, 275 Ma (Appendix S1, Figure S1.2). Our analyses support the origin of phenol production preceding benzoquinone production (Fig. 2A,C), and this chemical may be the first produced by millipede ancestors in the Silurian, and could be the oldest evidence of chemical production by land animals. Moreover, phenol production is the ubiquitous condition within Juliformia. The same pattern of phenol-benzoquinone evolution had been reported for Opiliones22 indicating that quinones could be produced by phenol para-oxidation and constitute —in many cases— an extension of the phenol biochemical pathway22,23. For millipedes it had been shown that phenol taxonomic distribution was patchy in Julida with multiple independent losses previously hypothesized24. Our reconstruction shows that phenol as a final product was lost multiple times in Juliformia (Fig. 2), and supports the hypothesis that phenol production is not essential for quinone production, further showing that other precursors may be involved in the pathway and that chemical complexity in millipedes was retained even in the absence of phenol production. Feeding experiments in millipedes have demonstrated that aromatic amino acids such as tyrosine may be precursors for phenol production mediated by tyrosine-phenol-lyase, an enzyme so far only recorded for Enterobacteriaceae48. Nonetheless, benzoquinone production in the absence of aromatic amino acids has been reported through the acetate pathway in millipedes and beetles with methionine as a precursor49,50. Even in the absence of phenols, the benzoquinone end product seems to require a hydroquinone precursor. This is supported by the presence of benzoquinones and their corresponding hydroquinones in the majority of our chemical samples. Our character reconstruction results show the ancestral condition for all Juliformia was the production of the precursor 2-methoxy-3-methylhydroquinone (Fig. 2A). Therefore, benzoquinone production originated by the ability to oxidize hydroquinones into their corresponding benzoquinone as had been hypothesized by earlier studies on arachnids23. The production of ethyl-benzoquinones is here only reported for Julidae, Rhinocricidae and Spirostreptidae, which are the three most chemically complex families. Recent analyses for cave-dwelling millipedes, demonstrate that ethyl-benzoquinone production had multiple independent origins, but it is not correlated to cave-dwelling21. Our results demonstrate independent origins even at the family level.
The retention of phenol production in Polydesmida lineages is not directly involved in the biosynthesis of their main aromatic defense chemicals (e.g., mandelonitrile, benzaldehyde, benzoyl cyanide), as these have been shown to derive from aromatic amino acids like phenylalanine51, nonetheless phenol has been proposed to be involved in maintaining the acidity needed for their biosynthesis52, and is the main chemical defense of immatures even when adults produce cyanogenics. This may suggest the secondary loss of cyanogenic production in some Polydesmida, probably due to the high cost of producing cyanogenics from amino acids53. Phenols are not directly involved in the production of other molecule groups such as quinazolinone alkaloids, or heterocyclic nitrogen-containing compounds20, which may indicate that these compounds arose as defenses independently.
Our results indicate that complexity arose from a simple ancestral condition of phenol production at least when Juliformia diverged from Polydesmida + Stemmiulida, and produced complex phenotypes convergently in three separate lineages, each from a different juliform order. Nonetheless, the ancestral complexity was also lost separately in two lineages. A central challenge in evolutionary biology is explaining complex evolutionary innovations that require the acquisition of multiple mutations. For metabolic innovations it has been shown that the addition of a single metabolic step in a pathway serves as a stepping stone towards the establishment of complex metabolic features in novel environments14. For millipedes, we have shown that independent lineages have gained similar levels of complexity independently, and the achievement of this complexity seems to have taken place in a step-wise manner. In the case of benzoquinones, the ability to produce phenols has been demonstrated to precede benzoquinone production and may have served as a stepping-stone for the production of hydroquinones, and these in place for the production of their corresponding benzoquinone. Even though these results are based on the most complete dataset to date, sampling is lower than ideal, considering the diversity of juliform lineages and chemistry. Nonetheless, the pattern that certain juliform families produce a considerably higher amount of chemicals than others is clear, and results so far support a gradual increase in complexity. Our study has major implications in the understanding of biodiversity, as adaptive traits greatly affect how taxa diversify through time. In response to predation, arthropods evolved novel defenses that enabled their escape from predators. These novel defenses facilitate species diversification among members of a clade sharing the same mechanism. After some time, predators develop the ability to adapt to these chemical defenses, and filling up empty niches, which allows for predator diversification. These predator-prey systems may produce a step-wise progression from simple to complex in the evolution of defense mechanisms54,55,56. For millipedes, an arms race with predators may have catalyzed the development of a metabolic stepping-stone process of evolutionary innovation. These novel biochemical defense secretion mechanisms potentially served as key innovations, allowing rapid diversification of the Juliformia and Polydesmida, which comprise approximately 75% of all nominal millipede species diversity in only four of 12 orders.
Materials and Methods
Transcriptomes were sequenced, and de novo assembled for 34 taxa across Diplopoda, then combined with 15 previously published transcriptomes (Appendix S1, Table S1.1). The resulting dataset was used to: (i) resolve the phylogenetic relationships between orders using 2,638 orthologs and divergence dates using fossil calibrations; (ii) investigate the origin and evolution of phenol and benzoquinone production; and (iii) determine the directionality of the evolution of complex chemical systems in the benzoquinone pathway. We identified shifts in the number of molecules produced where chemical complexity appears to have increased over time in some lineages and decreased in others. The composition of chemical defenses was determined through GC-MS analysis of secretions and existing chemical records.
RNA Extraction, Sequencing and Data Assembly
Total RNA was extracted from flash frozen or RNAlater preserved specimens using the TRIzol total RNA extraction method, purified with the RNeasy mini kit (Qiagen) and sequenced in-house at the Auburn University Core Genetics and Sequencing Laboratory using an Illumina Hi-Seq. 2500. Sequencing results (i.e. 100 bp paired end reads for each newly sequenced transcriptome) and fastq files from NCBI were de novo assembled in Trinity. New transcriptomes have been deposited into the NCBI Sequence Read Archive (SRA) (Appendix S1, Table S1.1). Resulting assemblies were analyzed in TransDecoder to produce predicted peptide sequences57.
Orthology Assessment
A custom core ortholog set was generated to infer orthologs in HaMStR v1358. The set was produced using transcriptomes from eight millipedes (Floridobolus sp., Sigmoria latior munda, Onomeris sp., Platydesmus sp., Polyxenidae sp., Pyrgodesmidae sp., Scoterpes sp.) and one centipede (Scutigera coleoptrata), which were chosen to cover a wide taxonomic sample. An all-versus-all BLASTP59 comparison was performed on the nine transcriptomes, with an e-value cut-off of 10−5, and used as input to perform Markov clustering in OrthoMCL 2.060 with an inflation parameter of 2.1. The putatively orthologous groups (OGs) produced were processed through the following steps: (1) remove sequences shorter than 100 amino acids in length; (2) align each candidate OG with MAFFT61 using the automatic alignment strategy and a maxiterate value of 1,000; (3) produce an “approximately maximum likelihood tree” for each OG with FastTree 262 using the “slow” and “gamma” options, by improving an initial neighbor-joining tree through minimum evolution with nearest neighbor interchange (NNI) subtree rearrangement, minimum evolution with subtree pruning regrafting (SPR) and maximum likelihood with NNI; (4) screen for paralogy using a tree-based approach in PhyloTreePruner63 by collapsing nodes with support values below 0.95, selecting a maximally inclusive tree (all taxa represented by no more than one sequence or more than one sequence forming a monophyletic group) and deleting putative paralogs (sequences falling outside of this maximally inclusive subtree) from the input alignment, where the longest sequence was retained for taxa with multiple sequences in a monophyletic group; and (5) discard all OGs with less than six of the nine taxa and/or not sampled for Sigmoria latior munda. These steps were performed using a modified version of the pipeline employed by Kocot et al.64. The pHMMs to be used in HaMStR were produced with hmmbuild and hmmcalibrate from the HMMER package65. Open reading frames produced by TransDecoder were searched against the 4,376 Millipede profile hidden Markov models (pHMMs), with Sigmoria latior munda —sampled in all OGs— as reference species. The steps outlined above were used to process the resulting putative OGs (modified from the Kocot et al.64 pipeline) and a supermatrix was produced (M1) with 2,638 orthologous sequence sets from 49 taxa by concatenation in FASconCAT66. Three reduced datasets were produced with 27% missing data (1250 orthologous sequence sets; M2), 21% missing data (625 orthologous sequence sets; M3), and 17% missing data (312 orthologous sequence sets; M4). We also calculated data informativeness for all partitions in PhyDesign67 and created two datasets with an informativeness score greater than 50 (M5) and the 312 most informative partitions (M6) for 30–200 Ma, focusing on resolving the position of Stemmiulida accurately (Appendix S1, Table S1.3).
Phylogenetic Analyses
For all matrices (M1-M6) an optimal maximum likelihood tree was obtained in the program ExaML version 3.0.168. Models of amino acid substitution were selected using the AUTOF command in ExaML. Node supports were estimated with 300 replicates generated and analyzed with the program RAxML and used to construct a majority-rule bootstrap consensus tree. A custom bash script (https://github.com/juanitarodrigueza/phylogenomics-pipeline/blob/master/bash_script) was used to automate the process and execute analyses on a high-performance computing (HPC) cluster. All analyses were conducted on the Auburn University CASIC HPS and Atrax (Bond Lab, Auburn University).
The program ASTRAL (Accurate Species TRee ALgorithm69) was used to infer a species tree from a series of unrooted gene trees. The ASTRAL approach works efficiently for large datasets and has been proposed to be more robust to incomplete lineage sorting, or deep coalescence, than maximum likelihood analysis of concatenated matrices. We first constructed individual gene trees for all partitions contained within matrices M1-M4. Maximum likelihood gene trees were generated in RAxML based on 100 random addition sequence replicates followed by 500 bootstrap replicates. Subsequent species tree estimation was inferred using ASTRAL v4.7.6, from all individual unrooted gene trees (and bootstrap replicates), under the multi-species coalescent model.
A reduced dataset with complete partitions for all taxa (M7; 38 partitions, 28 taxa) was generated for divergence time estimation in the program BEAST v1.8.170 under a uncorrelated lognormal relaxed clock model71,72. Two calibration schemes (C1, C2) were used for the analysis. For C1, the most recent common ancestor (MRCA) of Chilopoda was given a lognormal prior of (offset) 383 (μ = 2, SD = 0.5) based on the fossil Devonobius delta73. The MRCA of Diplopoda was given a uniform prior from 423 to 580 Ma. The lower bound was set based on the youngest age of the oldest diplopod fossil Pneumodesmus newmani deposits1 and the upper bound was set based on the oldest Ediacaran fossils, as a maximum hard bound for major arthropod clades74. The MRCA of Glomerida + Glomeridesmida was given a lognormal prior of (mean in real space) 33.9 Ma (SD = 2) based on the fossil Glomeris denticulata75. The MRCA of Julida was given a lognormal prior of (mean in real space) 33.9 (SD = 1) based on the fossil Parajulus cockerelli76. The MRCA of Nematophora (Callipodida + Chordeumatida) was given a uniform prior of lower 307 Ma upper 580 Ma. The lower bound is based on the earliest date of the deposit for the oldest fossil Nematophora genus Hexecontasoma77. The MRCA of the MRCA of Pyrgodesmidae was given a lognormal prior of (mean in real space) 13.65 based on the fossil Psochodesmus crescentis78. The model of protein evolution was determined for each partition using the perl script ProteinModelSelection.pl in RAxML. A template xml file was generated in BEAUti v.1.8.170 with all parameters and priors and each partition was transformed into an xml file using BEASTGen_v1.070. For each partition, four separate MCMC (Markov Chain Monte Carlo) searches were run until ESS values –assessed with LogAnalyser in BEAST- for all parameters were greater than 200. Tree files were combined for all runs in a single partition and 10,000 trees were randomly sampled. Trees from all partitions were combined using LogCombiner v1.7.5. A consensus tree was produced using TreeAnnotator v1.8.1. A custom bash script was used to process input files and run all analyses (https://github.com/juanitarodrigueza/phylogenomics-pipeline/blob/master/bash_script). For the second calibration method (C2) all the available fossil evidence was used to calculate the Penultimate Gap (the interval between the two oldest fossils in a clade; PenG) and the ghost lineage length (the difference between the oldest fossils for each of two sister lineages; GLin) as described by Norris et al. not in Reference section (http://biorxiv.org/content/biorxiv/early/2015/01/24/014340.full.pdf). These values were used to produce a prior distribution of the true age of crown-group Chilopoda and stem Helminthomorpha. The MRCA of crown-group Chilopoda was given a lognormal prior of (offset) 417 Ma (μ = 1.74, SD = 1,81). The MRCA of Helminthomorpha + Pentazonia was given a lognormal prior of (offset) 423 Ma (μ = 1.20,SD = 1.81). A chronogram containing all taxa was generated for ancestral character reconstruction using a penalized likelihood method using the chronos command in the R package ape47. The 95% highest posterior density dates obtained for the C2 BEAST analysis were incorporated as constraints for node ages.
Chemical Analysis
To collect the defensive compounds, live specimens were placed in a glass vial with 0.5 mL 100% methanol for a full body extraction. One specimen was sampled for each taxon. To avoid contamination, the glass vials had caps lined with Teflon. The methanol was analyzed using a Shimadzu QP-5000 GC/MS or Shimadzu QP-2010 GC/MS equipped with an RTX-5, 30 m × 0.25 mm i.d. column. The instruments were programmed from 60 °C to 250 °C at 10 °/min and held at that temperature for 20 min. To determine compounds, we used published data on mass spectra of quinone isomers. We also used and authentic samples of quinones as well as o-cresol, m-cresol, and p-cresol, present in our analytical chemistry facility. Retention indices were not used.
Ancestral Character Reconstruction
Chemical composition of defense secretion was mapped as a multistate character with states (1) No chemical, (2) Quinazolinone alkaloids, (3) Heterocyclic nitrogen-containing compounds, (4) Terpenes, (5) Benzoquinones and hydroquinones, (6) Phenol, (7) Cyanogenics. For the hypothetical phenol-benzoquinone pathway a single character was mapped with states (1) No chemical production, (2) Phenol production, (3) Benzoquinone production, and as a continuous character with states as number of chemical defense molecules found on hypothetical benzoquinone pathway79 (Fig. 2A), and assuming intermediate states as present (i.e. if a chemical found on the edge of the pathway was found, its hypothetical precursors were counted as present even if not reported in chemical results or literature) (see Supplemental Table S1.2, and S1.3). This character was scored at the family level, so that each terminal taxon represented the entire family lineage and its states corresponded to all chemical information available for the entire family. Discrete character states were mapped using a maximum likelihood approach implemented using the rayDISC command in the R package corHMM80. For chemical production, likelihood for the models of character evolution equal rates (ER), symmetrical (SYM) and all rates different (ARD) was calculated. The best model was determined using a likelihood ratio test. For phenol and benzoquinones we established a rate matrix that assumed different rates between transitions. This rate matrix assumed phenol as ancestral to benzoquinone as has been suggested for other arthropods22 and a single rate involved in losing the ability to produce one or all chemicals. The log-likelihood of this reconstruction was compared to that of other three models of character evolution: equal rates (ER), symmetrical (SYM) and all rates different (ARD). A likelihood-ratio test was performed to select among these varying models of character evolution. The model that best fit the data was the rate matrix model. The root state was determined using the procedure described by Maddison et al.81 and FitzJohn et al.82, treating the root state as a nuisance parameter and using an alternative root assignment that weights each root state according to its probability of giving rise to the extant data, given the model parameters and the tree. Continuous characters were mapped using the contMap command from the R package phytools83.
Data availability
Transcriptomes are available at NCBI Sequence Read Archive (SRA), assembled transcriptomes, ortholog alignments, and matrices are available from Dryad Data Repository.
Code availability
Scripts used in the analysis of these data are available at https://github.com/juanitarodrigueza.
References
Wilson, H. M. & Anderson, L. I. Morphology and taxonomy of paleozoic millipedes (Diplopoda: Chilognatha: Archipolypoda) from Scotland. J. Paleontol. 78, 169–184 (2004).
Selden, P. A. & Read, H. The oldest land animals: Silurian millipedes from Scotland. Bull. Br. Myriap. Isopod Gr. 23, 36–37 (2008).
Labandeira, C. C. Invasion of the continents: Cyanobacterial crusts to tree-inhabiting arthropods. Trends Ecol. Evol. 20, 253–262 (2005).
Anderson, D. P. et al. Evolution of an ancient protein function involved in organized multicellularity in animals. Elife 5, 1–20 (2016).
Marek, P. E. & Bond, J. E. A Müllerian mimicry ring in Appalachian millipedes. Proc. Natl. Acad. Sci. 106, 9755–9760 (2009).
Marek, P. E. & Moore, W. Discovery of a glowing millipede in California and the gradual evolution of bioluminescence in Diplopoda. Proc. Natl. Acad. Sci. 112, 6419–6424 (2015).
Skelhorn, J. & Ruxton, G. D. Ecological factors influencing the evolution of insects’ chemical defenses. Behav. Ecol. 19, 146–153 (2008).
Darwin, C. On the origin of species by means of natural selection. (Murray, 1859).
Nilsson, D. E. & Pelger, S. A pessimistic estimate of the time required for an eye to evolve. Proc. R. Soc. London B Biol. Sci. 256, 53–58 (1994).
Oakley, T. H. & Speiser, D. I. How complexity originates: The evolution of animal eyes. Annu. Rev. Ecol. Evol. Syst. 46, 237–260 (2015).
Sturmbauer, C., Levinton, J. S. & Christy, J. Molecular phylogeny analysis of fiddler crabs: test of the hypothesis of increasing behavioral complexity in evolution. Proc. Natl. Acad. Sci. USA 93, 10855–10857 (1996).
Finnigan, G. C., Hanson-Smith, V., Stevens, T. H. & Thornton, J. W. Evolution of increased complexity in a molecular machine. Nature 481, 360–364 (2012).
Liu, R. & Ochman, H. Stepwise formation of the bacterial flagellar system. Proc. Natl. Acad. Sci. 104, 7116–7121 (2007).
Szappanos, B. et al. Adaptive evolution of complex innovations through stepwise metabolic niche expansion. Nat. Commun. 7, 11607 (2016).
Brewer, M. S. & Bond, J. E. Ordinal-level phylogenomics of the arthropod class Diplopoda (millipedes) based on an analysis of 221 nuclear protein-coding loci generated using next-generation sequence analyses. PLoS One 8, e79935 (2013).
Brewer, M. S., Sierwald, P. & Bond, J. E. Millipede taxonomy after 250 years: classification and taxonomic practices in a mega-diverse yet understudied arthropod group. PLoS One 7, e37240 (2012).
Crawford, C. S. Millipedes as model detritivores. Berichte des Naturwissenschaftlich-Medizinischen Vereins Innsbruck Suppl. 10, 277–288 (1992).
Smit, A. M. & Van Aarde, R. J. The influence of millipedes on selected soil elements: A microcosm study on three species occurring on coastal sand dunes. Funct. Ecol. 15, 51–59 (2001).
Sierwald, P. & Bond, J. E. Current status of the Myriapod class Diplopoda (millipedes): taxonomic diversity and phylogeny. Annu. Rev. Entomol. 52, 401–420 (2007).
Shear, W. A. The chemical defenses of millipedes (Diplopoda): Biochemistry, physiology and ecology. Biochem. Syst. Ecol. 61, 78–117 (2015).
Makarov, S. E. et al. Chemical ecology of cave-dwelling millipedes: Defensive secretions of the Typhloiulini (Diplopoda, Julida, Julidae). J. Chem. Ecol. 43, 317–326 (2017).
Raspotnig, G. et al. Chemosystematics in the Opiliones (Arachnida): A comment on the evolutionary history of alkylphenols and benzoquinones in the scent gland secretions of Laniatores. Cladistics 31, 202–209 (2015).
Rocha, D. F. O. et al. Harvestman phenols and benzoquinones: Characterisation and biosynthetic pathway. Molecules 18, 11429–11451 (2013).
Bodner, M. et al. ‘Quinone millipedes’ reconsidered: Evidence for a mosaic-like taxonomic distribution of phenol-based secretions across the Julidae. J. Chem. Ecol. 42, 249–258 (2016).
Shear, W. A. & Edgecombe, G. D. The geological record and phylogeny of the Myriapoda. Arthropod Struct. Dev. 39, 174–190 (2010).
Rehm, P., Meusemann, K., Borner, J., Misof, B. & Burmester, T. Phylogenetic position of Myriapoda revealed by 454 transcriptome sequencing. Mol. Phylogenet. Evol. 77, 25–33 (2014).
Fernández, R., Edgecombe, G. D. & Giribet, G. Exploring phylogenetic relationships within Myriapoda and the effects of matrix composition and occupancy on phylogenomic reconstruction. 65, 871–889 (2015).
Fernández, R., Edgecombe, G. D. & Giribet, G. Phylogenomics illuminates the backbone of the Myriapoda Tree of Life and reconciles morphological and molecular phylogenies. Sci. Rep. 8, 83 (2018).
Enghoff, H. Phylogeny of millipedes- a cladistic analysis. Zool. Syst. Und Evol. 22, 8–26 (1984).
Sierwald, P., Shear, W. A., Shelley, R. M. & Bond, J. E. Millipede phylogeny revisited in the light of the enigmatic order Siphoniulida. J. Zool. Syst. Evol. Res. 41, 87–99 (2003).
Verhoeff, K. W. Klasse Diplopoda. Bronns Klassen und Ordungen der Tier-Reichs 5, I–XII, I–VI, 1–2084 (1926).
Manton, S. M. The evolution of arthropodan locomotory mechanisms-part 5: The structure, habits and evolution of the Pselaphognatha (Diplopoda). J. Linn. Soc. London, Zool 43, 153–187 (1957).
Boudreaux, H. B. In Arthropod phylogeny (ed. Gupta, A. P.) 551–586 (Van Nostrand Reinhold Company, 1979).
Enghoff, H., Dohle, W. & Blower, J. G. Anamorphosis in millipedes (Diplopoda): the present state of knowledge with some developmental and phylogenetic considerations. Zool. J. Linn. Soc. 109, 103–234 (1993).
Verhoeff, K. W. Die Ordnungen der Proterandria und zur Kenntnis der Cambaliden (Über Diplopoden. 65. Aufsatz.). Zool. Anz. 43, 49–65 (2003).
Hoffman, L. Classification of the Diplopoda. (Muséum d’Histoire Naturelle, Genève, 1980).
Golovatch, S. I. A review of the volvatory Polydesmida, with special reference to the patterns of volvation (Diplopoda). African Invertebr. 44, 39–60 (2003).
Blanke, A. & Wesener, T. Revival of forgotten characters and modern imaging techniques help to produce a robust phylogeny of the Diplopoda (Arthropoda, Myriapoda). Arthropod Struct. Dev. 43, 63–75 (2014).
Edgecombe, G. D. In Treatise on Zoology- Anatomy, Taxonomy, Biology- The Myriapoda (ed. Minelli, A.) 353–362 (Brill Academic Publishers, 2015).
Hopkins, T. L. & Kramer, K. J. Insect cuticle sclerotization. Annu. Rev. Entomol. 37, 273–302 (1992).
Verhoeff, K. W. Die Ordnungen der Proterandria und zur Kenntnis der Cambaliden (Über Diplopoden. 65. Aufsatz.). Zool. Anz. 43, 49–65 (1913).
Shear, W. A. et al. Early land animals in North America: Evidence from Devonian age arthropods from Gilboa, New York. Science (80) 224, 492–494 (1984).
Shear, W. A. Spinnerets in the milliped order Polydesmida, and the phylogenetic significance of spinnerets in millipeds (Diplopoda). Int. J. Myriap. 2, 123–146 (2008).
Xi, Z., Liu, L. & Davis, C. C. The impact of missing data on species tree estimation. Mol. Biol. Evol. 33, 838–860 (2015).
Enghoff, H. In Treatise on Zoology- Anatomy, Taxonomy, Biology- The Myriapoda (ed. Minelli, A.) 427–428 (Brill Academic Publishers, 2015).
Sanderson, M. Estimating absolute rates of molecular evolution and divergence times: A penalized likelihood approach. Mol. Biol. Evol. 19, 101–109 (2002).
Paradis E., Claude J. & Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (2004).
Duffey, S. S. & Blum, M. S. Phenol and guaiacol: Biosynthesis, detoxication, and function in a polydesmid millipede, Oxidus gracilis. Insect Biochem. 7, 57–65 (1977).
Meinwald, J., Koch, K. F., Rogers, J. E. J. & Eisner, T. Biosynthesis of arthropod secretions. III Synthesis of simple p-benzoquinones in a beetle (Eleodes longicollis). J. Am. Chem. Soc. 88, 1590–1592 (1966).
Blum, M. S. Biosynthesis of arthropod exocrine compounds. Annu. Rev. Entomol. 32, 381–413 (1987).
Towers, G. H. N., Duffey, S. S. & Siegel, S. M. Defensive secretion: biosynthesis of hydrogen cyanide and benzaldehyde from phenylalanine by a millipede. Can. J. Zool. 50, 1047–1050 (1972).
Ishida, Y. et al. A sacrificial millipede altruistically protects its swarm using a drone blood enzyme, mandelonitrile oxidase. Sci. Rep. 6, 26998 (2016).
Kuwahara, Y., Ichiki, Y., Morita, M., Tanabe, T. & Asano, Y. Chemical polymorphism in defense secretions during ontogenetic development of the millipede Niponia nodulosa. J. Chem. Ecol. 41, 15–21 (2014).
Boevé, J.-L., Blank, S. M., Meijer, G. & Nyman, T. Invertebrate and avian predators as drivers of chemical defensive strategies in tenthredinid sawflies. BMC Evol. Biol. 13, 198 (2013).
McGlothlin, J. W. et al. Historical contingency in a multigene family facilitates adaptive evolution of toxin resistance. Curr. Biol. 12, 1616–1621 (2016).
Zaman, L. et al. Coevolution Drives the Emergence of Complex Traits and Promotes Evolvability. PLoS Biol 12, e1002023 (2014).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat. Protoc. 8, https://doi.org/10.1038/nprot.2013.084 (2013).
Ebersberger, I., Strauss, S. & von Haeseler, A. HaMStR: Profile hidden markov model based search for orthologs in ESTs. BMC Evol. Biol. 9, 157 (2009).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Katoh, K., Kuma, K., Toh, H. & Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518 (2005).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – Approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010).
Kocot, K. M., Citarella, M. R., Moroz, L. L. & Halanych, K. M. PhyloTreePruner: A phylogenetic tree-based approach for selection of orthologous sequences for phylogenomics. Evol. Bioinform. Online 9, 429–435 (2013).
Kocot, K. M. et al. Phylogenomics reveals deep molluscan relationships. Nature 477, 452–456 (2011).
Eddy, S. R. Accelerated Profile HMM Searches. PLoS Comput Biol 7, e1002195 (2011).
Kück, P. & Meusemann, K. FASconCAT: Convenient handling of data matrices. Mol. Phylogenet. Evol. 56, 1115–1118 (2010).
López-Giráldez, F. & Townsend, J. P. PhyDesign: An online application for profiling phylogenetic informativeness. BMC Evol. Biol. 11, 152 (2011).
Kozlov, A. M., Aberer, A. J. & Stamatakis, A. ExaML version 3: A tool for phylogenomic analyses on supercomputers. Bioinformatics 31, 2577–2579 (2015).
Mirarab, S. et al. ASTRAL: Genome-scale coalescent-based species tree estimation. Bioinformatics 30, i541–i548 (2014).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 8, 1969–1973 (2012).
Drummond, A. J., Ho, S. Y. W., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol 4, e88 (2006).
Drummond, A. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Shear, W. A. & Bonamo, P. M. Devoniomorpha, a new order of centipeds (Chilopoda) from the Middle Devonian of Gilboa, New York State, USA, and the phylogeny of centiped orders. Am. Museum Novit. 2927, 1–30 (1988).
Misof, B. et al. Phylogenomics resolves the timing and pattern of insect evolution. Science (80-.). 346, 763–767 (2014).
Menge, A. Footnotes and taxonomic names, in Die im Bernstein befindlichen Crustaceen, Myriapoden, Arachniden und Apteren der Vorwelt. Die Bernstein Befindlichen Org. Reste der Vor. Gesammelt Verbindung mit Mehreren Bearbeitetet und Hrsg. 1, 1–124 (1854).
Miner, R. A fossil myriapod of the genus Parajulus from Florissant, Colorado. Am. Museum Novit. 219, 1–5 (1926).
Hannibal, J. Hexecontasoma, a new helminthomorph millipede (Hexecontasomatidae n. fam.) from the Mazon Creek, Illinois, fauna (Carboniferous, North America. Fragm. Faun 43, 19–35 (2000).
Loomis, H. The millipeds of Hispaniola, with descriptions of a new family, new genera, and new species. Bull. Museum Comp. Zool. 80, 1–191 (1936).
Deml, R. & Huth, A. Benzoquinones and hydroquinones in defensive secretions of tropical millipedes. Naturwissenschaften 87, 80–82 (2000).
Beaulieu, J. M., Oliver, J. C. & O’Meara, B. CorHMM: Analysis of binary character evolution (2016).
Maddison, W. P., Midford, P. E. & Otto, S. P. Estimating a binary character’s effect on speciation and extinction. Syst. Biol. 56, 701–710 (2007).
FitzJohn, R. G., Maddison, W. P. & Otto, S. P. Estimating trait-dependent speciation and extinction rates from incompletely resolved phylogenies. Syst. Biol. 58, 595–611 (2009).
Revell, L. J. phytools: an R package for phylogenetic comparative biology (and other things). 3, 217–223 (2011).
Acknowledgements
This work was supported by National Science Foundation grant DEB 1256139 to J.E.B., P.S., W.A.S., T.H.J. and P.E.M.
Author information
Authors and Affiliations
Contributions
J.E.B., P.S., W.A.S., J.R., M.E.B., T.H.J. and P.E.M. devised the project, J.E.B., P.E.S., P.E.M. and W.H.S. obtained funding, J.E.B. and P.E.S. administered funds, J.E.B. and J.R. gathered and analyzed all data except for core ortholog sets which were obtained by K.M.K., J.E.B. and J.R. wrote the first draft and prepared the figures, T.H.J. analyzed chemical samples, J.E.B., P.S., W.A.S., J.R., M.E.B., and P.E.M. collected specimens, J.E.B., P.E.S., P.E.M., W.A.S., T.H.J., J.R., K.M.K., and M.E.B. read and edited the first draft.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rodriguez, J., Jones, T.H., Sierwald, P. et al. Step-wise evolution of complex chemical defenses in millipedes: a phylogenomic approach. Sci Rep 8, 3209 (2018). https://doi.org/10.1038/s41598-018-19996-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-19996-6
This article is cited by
-
Alkaloid chemistry in pill-millipedes: Defensive secretion in two species of Typhloglomeris Verhoeff, 1898 (Diplopoda, Glomerida, Glomeridellidae)
Chemoecology (2024)
-
De novo metatranscriptomic exploration of gene function in the millipede holobiont
Scientific Reports (2022)
-
The first true millipede—1306 legs long
Scientific Reports (2021)
-
Attraction of Canthon vazquezae (Coleoptera: Scarabaeinae) to Volatiles Released by Messicobolus magnificus (Diplopoda: Spirobolida)
Journal of Insect Behavior (2021)
-
Shifting evolutionary sands: transcriptome characterization of the Aptostichus atomarius species complex
BMC Evolutionary Biology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
