
Abstract
Isoprene is the most abundant non-methane volatile organic compound (VOC) and the largest contributor to secondary organic aerosol (SOA) burden on a global scale. In order to examine the influence of high concentrations of anthropogenic pollutants on isoprene-derived SOA (SOA i ) formation, summertime PM2.5 filter samples were collected with a three-hour sampling interval at a rural site in the North China Plain (NCP), and determined for SOA i tracers and other chemical species. RO2+NO pathway derived 2-methylglyceric acid presented a relatively higher contribution to the SOA i due to the high-NOx (~20 ppb) conditions in the NCP that suppressed the reactive uptake of RO2+HO2 reaction derived isoprene epoxydiols. Compared to particle acidity and water content, sulfate plays a dominant role in the heterogeneous formation process of SOA i . Diurnal variation and correlation of 2-methyltetrols with ozone suggested an important effect of isoprene ozonolysis on SOA i formation. SOA i increased linearly with levoglucosan during June 10–18, which can be attributed to an increasing emission of isoprene caused by the field burning of wheat straw and a favorable aqueous SOA formation during the aging process of the biomass burning plume. Our results suggested that isoprene oxidation is highly influenced by intensive anthropogenic activities in the NCP.
Similar content being viewed by others
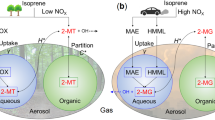


Introduction
Atmospheric secondary organic aerosols (SOA) are produced from photochemical oxidation products of volatile organic compounds (VOCs) via gas-particle conversion processes such as nucleation, condensation and heterogeneous chemical reactions1. SOA account for about 70 percent of global organic aerosols in the troposphere1, and have important impacts on climate and human health2,3,4,5. Among all the precursors isoprene is the most important contributor to the global SOA burden6,7. Annual isoprene emission on the Earth surface is about 600 Tg yr−1 8, comprising approximately half of the total VOC emissions from both natural and anthropogenic sources7,9. On a global scale, isoprene-derived products is about 19.2 TgC yr−1, accounting for ~70% of the total SOA10.
A number of studies found that oxidation of isoprene by hydroxyl radical (OH) generates large amounts of aerosol phase low-volatility products6,11,12,13. Under low-NOx conditions isoprene reacts with OH and HO2 radicals and produces isoprene epoxydiols (IEPOX)12,14,15, while under high-NOx conditions isoprene reacts with OH radical and NOx, producing methacrylic acid epoxide (MAE)16 and hydroxymethyl-methyl-lactone (HMML)17. Those epoxides are critical intermediates of isoprene SOA formation through reactive uptake by acidic particles. In addition, recent studies found that ozonolysis of isoprene leads to stabilized Criegee intermediates (sCIs), which could also be an important contributor to isoprene-derived SOA (SOA i ) in the atmosphere18,19. Moreover, many studies have found that relative humidity (RH)20,21, aerosol acidity22,23,24, anthropogenic pollutants such as gaseous NOx and SO225,26,27 and particulate sulfate13,14 have pronounced effects on SOA i formation.
In comparison with other countries, the atmospheric environment of China has been suffering from high levels of SO2, NOx, O3, VOCs, NH3 and particulate matter (PM) especially in North China Plain (NCP). Due to the lack of efficient emission controls during the fast industrialization and urbanization processes, severe haze episodes frequently occurred in the region in the past decades28,29. In addition, field open burning is still a common activity for disposal of crop residue in the rural area of NCP, especially in summertime wheat harvest period, which releases huge amount of pollutants into the atmosphere and leads to significant impacts on the air quality and aerosol properties30,31,32.
Many previous studies in the NCP focused on anthropogenic source distributions of gaseous and particle pollutants, and their environmental, ecological and human health effects33,34,35,36. However, only few studies examined how biogenic SOA is affected by such high loadings of anthropogenic pollutants in the region37,38,39. In the current study, PM2.5 filter samples were collected with a three-hour sampling interval at a rural site in the central part of NCP during June 10th to 25th, 2013, and determined for SOA i tracers to explore the influence of anthropogenic pollution on SOA i formation. We firstly investigated the temporal variation and chemical composition of SOA i in NCP, and then examined the impacts of sulfate, ozone and biomass burning on the SOA i formation.
Results and Discussion
Temporal variation of isoprene-derived SOA tracers
3-Methyltetrahydrofuran-3,4-diols (3-MeTHF-3,4-diols), 2-methylglyceric acid, C5-alkene triols and 2-methyltetrols were detected in all the filter samples collected at the Gucheng Meteorological Station (Table 1). Among the eight isoprene-derived SOA tracers, 3-MeTHF-3,4-diols, C5-alkene triols and 2-methyltetrols are mainly formed by reactive uptake of IEPOX, key intermediates that are produced by RO2+HO2 reactions of isoprene without involvement of NOx12,14. The gas-phase IEPOX can be trapped into the aerosol phase through acid-catalyzed intermolecular rearrangement reactions to form 3-MeTHF-3,4-diols and/or C5-alkene triols40,41. Thus, 3-MeTHF-3,4-diols and C5-alkene triols at Gucheng presented a similar temporal variation trend (Fig. 1c) with a strong linear correlation (r2 = 0.91, Figure S2a). 2-Methyltetrols are primarily formed through the nucleophilic addition of water to the ring-opened products of the epoxydiols in aerosol phase12, which is a more complex pathway compared to 3-MeTHF-3,4-diols and C5-alkene triols formation. Moreover, Riva et al.18 recently demonstrated that isoprene ozonolysis could be another source for 2-methyltetrols in the presence of acidic sulfate aerosol (detailed mechanism discussed in the following section). Thus it is reasonable that correlation of 2-methyltetrols with 3-MeTHF-3,4-diols (r2 = 0.66) or C5-alkene triols (r2 = 0.54) (Figs 1 and S2b) was not as strong as 3-MeTHF-3,4-diols and C5-alkene triols.

Temporal variations of biogenic secondary organic aerosols (BSOA) derived from isoprene, temperature, relative humidity, pHIS (represents particle acidity), LWC (liquid water content), OC and EC (organic and elemental carbon). MeTHFdiols: the sum of cis-3-methyltetrahydrofuran-3,4-diol and trans-3-methyltetrahydrofuran-3,4-diol; C5-alkene triols: the sum of 3-methyl-2,3,4-trihydroxy-1-butene, cis-2-methyl-1,3,4-trihydroxy-1-butene, and trans-2-methyl-1,3,4-trihydroxy-1-butene; 2-MTs: the sum of 2-methylthreitol and 2-methylerythritol; 2-MGA: 2-methylglyceric acid.
Under high-NOx conditions isoprene can be oxidized into methacrolein (MACR) through RO2+NO reaction, which undergoes further oxidation with OH and NOx radicals to form methacryloyl peroxynitrate (MPAN)25,42. MPAN may react with OH radical to produce MAE and HMML. Those two compounds undergo further reactions under acid conditions, forming 2-methylglyceric acid and some related dimers, organic nitrates and organosulfates17,27. Field studies conducted at low-NOx sites (0.1–2 ppb) in the US27,43,44 and Germany45 showed that 2-methylglyceric acid concentrations are almost one order of magnitude lower than 2-methyltetrols and C5-alkene triols. In contrast, NOx levels (around 20 ppb)33 in NCP are much higher. Thus, the role of RO2+NO pathway in SOA i formation is more important in the region. As shown in Table 1, concentrations of 2-methylglyceric acid (2.0–60 ng m3, ave.19 ng m3) in the PM2.5 samples at the Gucheng site are only 2–4 times lower than those of 2-methyltetrols (4.0–134 ng m3, ave. 45 ng m3) and C5-alkene triols (4.3–219 ng m3, ave. 41 ng m3). Coincidentally, a recent real-time observation in Eastern China (Nanjing) revealed a suppression of IEPOX-SOA formation caused by the low IEPOX reactive uptake rate and NO-dominant gas-phase chemistry under high-NOx conditions (21.4 ppb)46. Both results highlight the important role of the severe anthropogenic pollution in isoprene-derived SOA formation process in the NCP atmosphere.
All the SOA i tracers showed a similar temporal variation pattern to organic carbon (OC) (Fig. 1e); concentration of total SOA i tracers linearly correlated with that of OC with a coefficient of r2 = 0.61, suggesting that isoprene oxidation is an important source of organic aerosols in the rural area of NCP.
Influence of sulfate, aerosol acidity and water content
Acid-catalyzed reactive uptake and subsequent particle-phase reactions of IEPOX (for 3-MeTHF-3,4-diols, C5-alkene triols and 2-MTs) and MAE or HMML (for 2-MGA) are the most important chemical pathways of SOA formation from isoprene12,16. Several previous studies found that aerosol particles of varying acidity and liquid water contents can affect the SOA i yield22,23,47,48. In order to examine the aerosol aqueous chemistry during the SOA i formation, ISORROPIA-II, a thermodynamic model, was performed to estimate the aerosol acidity (i.e., In-situ pH, pH IS , or H+ concentration in the aqueous phase, H+aq) and liquid water content (LWC). As mentioned in Methods section, pH values could approximately be underestimated by one unit due to the lack of NH3 data49,50. In the current study, pH values of PM2.5 ranged from 0.2 to 4.1 with an average of 2.1 ± 0.6 at the Gucheng site (Fig. 1b), indicating that the acidity of aerosol in the rural region of NCP is much weaker than that in southeast US (0.5–2.0)49, mainly due to the abundant alkaline NH3 in the region29. In contrast, aerosol water content (42 ± 50 μg m−3) at the Gucheng site is about 10 times higher than that (5.1–8.4 μg m−3) in southeastern US48, which can be ascribed to the high loading of inorganic ions in NCP.
Based on a multivariate linear regression analysis of SOA i tracers (r2 = 0.41, p < 0.001) with sulfate, particle acidity (represented as H+aq), and water content, we found that only sulfate has a statistically significant (p < 0.001) positive linear relationship with SOA i tracers (Table S1). Moreover, Fig. 2 showed that all the detected SOA i tracers present moderate or good linear correlation (r2 = 0.3~0.6) with sulfate concentration, again demonstrating the enhancing effect of sulfate on SOA i formation, which is consistent with recent studies in the southeast US43,44,48,51. Such an effect can be primarily explained by two reasons: 1) acidic sulfate provides a surface that is favorable for acid-catalyzed reactive uptake and ring-opening reaction of the key intermediates in the gas phase14,43, 2) salting-in effect of sulfate increases the solubility of polar organic compounds like IEPOX, MAE and HMML48. However, as shown in Fig. 2, we found that sulfate and SOA i tracers present stronger correlations and higher slopes in lower acidity conditions (pH > 2 in the current study, the red dots in Fig. 2), which indicates that reactive acidic uptake of SOA i precursors is more responsible for sulfate growth in the atmosphere when aerosols acidity is weak. This phenomenon may be related to the effects of sulfate on IEPOX reaction probability (γIEPOX), aerosol acidity52,53, surface area54, and the exact explanation needs further research.

Relationships between sulfate and determined isoprene-derived products.
In agreement with the multivariate regression analysis, no significant correlations of the detected SOA i tracers with particle acidity and liquid water content (LWC) of aerosol were found in the current study (Fig. 1b), consisting with the results observed in southeastern US43,48. This is because that the influences of aerosol acidity and water content on isoprene oxidation are very complicated processes in ambient environment, which can also involve in viscosity or morphology changes and liquid–liquid phase separations of aerosols, dilution effects on ion strength and acidity by particle-phase water, and competitions from gas phase oxidation48,55. In addition, particle acidity and liquid water content may vary regionally, which can also influence their relationships to SOA i concentration in the sampling site when SOA are formed during a long-distance transport process.
Influence of ozone
Figure 3 displays diurnal variations of the SOA i tracers at Gucheng station during June 11–19, 2013, which was a period without precipitation. Both 3-MeTHF-3,4-diols and C5-alkene triols presented higher concentrations in the nighttime than in the daytime (Fig. 3a and b) with a statistic significance (p < 0.05). The planetary boundary layer (PBL) height decrease (Fig. 3d) due to the nighttime lower temperature (Fig. 1) would be the primary reason. On the other hand, as showed in Fig. 3c, higher concentration of sulfate can enhance the yield of isoprene-derived SOA by promoting gas-aerosol phase conversion of its precursors and the subsequent aqueous phase chemistry. Thus, the diurnal variation of both 3-MeTHF-3,4-diols and C5-alkene triols showed a maximum at 21:00–24:00 p.m. and a minimum in the afternoon (12:00–15:00 p.m.).

Diurnal variation of the determined isoprene-derived products, sulfate, ozone and Planetary Boundary Layer (PBL) height (data provided by Real-time Environmental Applications and Display System, https://ready.arl.noaa.gov/READYamet.php) during June 11–19.
However, 2-methyltetrols showed a diurnal variation pattern different from that of 3-MeTHF-3,4-diols and C5-alkene triols. Concentrations of 2-methyltetrols displayed an obvious increase in the afternoon, peaking at 15:00–18:00 p.m. In addition to •OH-initiated oxidation, isoprene ozonolysis is also a potential pathway of SOA formation56. Previous studies19,57 proposed that initial formation of isoprene primary ozonides leads to stabilized sCIs, which can further react in the gas phase to form higher molecular weight products that subsequently partition to the aerosol phase and form SOA. Based on their chamber study, Riva et al.18 found that in the presence of wet acidic aerosols isoprene ozonolysis yields 2-methyltetrols and organosulfates but not produces C5-alkene triols, 3-MeTHF-3,4-diols and 2-methylglyceric acid. They tentatively proposed that hydroperoxides formed in the gas phase from isoprene ozonolysis potentially partition to wet acidic sulfate aerosols and hydrolyze to 2-methyltetrols and related organosulfates. Recently, Rattanavaraha et al.44 verified that isoprene ozonolysis plays a role in 2-methyltetrols formation process. During the current study period, ozone showed a highest concentration in the afternoon (12:00–15:00 p.m.) (Fig. 3d), and weakly correlated with 2-methyltetrols (r2 = 0.21, Fig. 4a) in the daytime, suggesting an important role of isoprene ozonolysis in the SOA formation process in NCP. Additionally, the non-correlation of ozone with 3-MeTHF-3,4-diols and C5-alkene triols and the different diurnal variation patterns of those SOA i tracers suggest that isoprene ozonolysis would have limited influence on these compounds.

Relationships of (a) 2-methylthetrols and (b) 2-methylglyceric acid with ozone during the whole sampling period, and total determined isoprene-derived products with levoglucosan during (c) June 10~18 and (d) June 19–25.
In consistent with the field study by Rattanavaraha et al.44, 2-methylglyceric acid, represented as MAE/HMML-derived SOA, also presented a moderate correlation (r2 = 0.34, Fig. 4b) with O3 concentration during the daytime, and was highest during 15:00–18:00 p.m., because the formation of MACR (a precursor to MAE and HMML) was enhanced by oxidation of isoprene by O344.
Influence of regional biomass burning
From the end of May to the mid of June is the wheat harvest season and field open burning of wheat straw is a common activity in the rural area of NCP. Based on the NASA satellite observation of fire spots (https://firms.modaps.eosdis.nasa.gov/firemap/) (Fig. 5a), we found that intensive emissions from the wheat straw burning occurred during the first half of the sampling period (i.e., June 10–18, 2013) in the NCP region. As shown in Fig. 5b, 77% of the air masses arriving at the Gucheng station during June 10–18 were transported via long distances from the southeast part of NCP. Some emission inventory studies revealed that biomass burning could release large amounts of isoprene to the atmosphere58,59. Zhu et al.60 examined the amounts of VOCs in the air at another rural site (Yucheng, Shandong Province, China) in the NCP region, and found that isoprene concentration during the wheat straw burning period is around double of that in normal periods. Thus, in order to discuss the influence of the regional intense burning of biomass fuels on isoprene-derived SOA in the rural areas of NCP, the relationships between SOA i tracers and biomass burning tracer (i.e., levoglucosan) were analyzed (Fig. 4c and d). During June 10–18, the detected SOA i tracers positively correlated with levoglucosan with a high coefficient (r2 = 0.67 in the daytime and 0.68 in the nighttime) (Fig. 4c), suggesting that emissions from the biomass burning enhanced the isoprene-derived SOA formation in the NCP region.

Fire spots (a,c) (provided by Fire Information for Resource Management System, FIRMS, https://firms.modaps.eosdis.nasa.gov/firemap/, Collection 6 and MCD14ML), and cluster analyses of air masses (b,d) (original data of backward trajectories provided by NOAA HYSPLIT modeling system68,69, http://ready.arl.noaa.gov/HYSPLIT.php) during the sampling period (June 10–18 and June 19–25, 2013). Sampling site represented as red star.
As shown in Fig. 5c and d, during the late half of sampling period (i.e., June 19–25), the number of fire spots decreased sharply, suggesting that field burning biomass during the period was insignificant. Thus the correlation between the SOA i tracers and levoglucosan during the period was not as significant as that during June 10–18 (Fig. 4d). However, the SOA i tracers and levoglucosan still had a moderate relationship (r2 = 0.52) in the nighttime during June 19–25. As shown in Fig. 5d, most air masses (62%) of Gucheng during the late half period were transported from the local area. Because at high isoprene emission from natural plants is insignificant and the local residential biomass burning is the major source, thus the correlation of the SOA i tracers with levoglucosan was only observed at night. Interestingly, the nighttime SOA i tracer concentrations during June 10–18 (150 ± 115 ng m−3) were obviously higher than those in the period of June 19–25 (85 ± 53 ng m−3) (Table S2), although the nighttime concentrations of levoglucosan during the two phases (250 ± 182 and 218 ± 208 ng m−3) were comparable. Recently, Gilardoni et al.61 directly observed a strong formation of aqueous secondary organic aerosol from biomass burning emissions. At the Gucheng site nighttime concentration (26 ± 16 μg m−3) of sulfate during June 10–18 was more than 2 times higher than that (12 ± 11 μg m−3) in the remaining period (Table S2), which can be attributed to a continuous aqueous oxidation of SO2 from biomass burning and other anthropogenic activities during the long-range transport at the first period. As discussed in Section 3.2, sulfate plays an important role in promoting aqueous phase oxidation of isoprene-derived products. Thus, the higher SOA i concentration during the nighttime of June 10–18 was attributed to an enhancing aqueous-phase chemistry of isoprene-derived organics during the biomass burning smoke transport process.
Methods
Field Sampling
The measurement was performed at the Integrated Ecological-Meteorological Observation and Experiment Station of Chinese Academy of Meteorological Sciences (39°08′N, 115°40′E, 15.2 m asl, Figure S1), which is located in a rural area of Gucheng, Hebei Province. Detailed information of the station and some observation results of meteorological condition and air quality in the area were described elsewhere33. Briefly, the Gucheng site is in the central zone of Beijing-Tianjin-Hebei Region, about 110 km southwest to Beijing (See Figure S1). Taihang Mountains, with a range extending over 400 km from north to south, is about 30 km away from the west of the site. The sampling station is surrounded by the farms and villages, where the main crops are corn and wheat in summer. Weather conditions during the observation period are characteristic of warm and humid (24 ± 4 °C and 67 ± 19%). In all the seasons, the prevailing surface wind directions were northeasterly and southwesterly. A previous study showed that the concentrations of NOx, SO2, and O3 in June (2007) were around 20 ppb, 10 ppb, and 50 ppb, respectively, indicating severe air pollution in summer in the region33.
PM2.5 samples were collected on the rooftop (about 10 m above the ground) of a three-story building at the site of the Gucheng station. The sampling was conducted from June 10th to 25th, 2013 by using a high volume (1.13 m3 min−1) sampler (Anderson) with a three-hour sampling interval. A total of 118 field samples were collected onto pre-baked (450 °C, 6–8 hr) quartz fiber filters (20.3 cm * 25.4 cm). Three field blank samples were also collected by mounting blank filters onto the sampler for about 15 min without pumping any air. After sampling, the sample filter was individually sealed in pre-baked aluminum foil bags and stored in a freezer (−20 °C) prior to analysis.
Chemical analysis
A punch of the filter (including both field and blank samples, N = 121) with an area of 12.5–25 cm2 was extracted three times with a mixture of dichloromethane and methanol (2:1, v/v) each for 10 min under ultrasonication. The extracts were concentrated using a rotary evaporator under vacuum conditions and then blow down to dryness using pure nitrogen. After reaction with 50 μL N,O-bis-(trimethylsilyl) trifluoroacetamide (BSTFA) with 1% of trimethylsilyl chloride and 10 μL of pyridine at 70 °C for 3 hr, the derivatives were determined using gas chromatography/electron ionization mass spectrometry (GC/EI-MS)55.
GC/EI-MS analysis of the derivatives was performed using an Agilent 7890 A GC coupled with an Agilent 5975 C MSD. GC oven temperature program and MS conditions were described elsewhere55,62,63. Individual characteristic ions, m/z 262 for 3-MeTHF-3,4-diols (trans-3-methyltetrahydrofuran-3,4-diol and cis-3-methyltetrahydrofuran-3,4-diol), m/z 219 for 2-MGA (2-methylglyceric acid), m/z 231 for C5-alkene triols (cis-2-methyl-1,3,4-trihydoxy-1-butene, 3-methyl-2,3,4-trihydoxy-1-butene and trans-2-methyl-1,3,4-trihydoxy-1-butene), and m/z 219 for 2-MTs (2-methylthreitol and 2-methylerythritol), are used for quantification of the isoprene-derived SOA tracers. However, the response factors of all the determined products are replaced by erythritol, because the authentic standards are not commercially available27,37. No significant contamination (<5% of those in the samples) was found in the blanks. In addition, it is worth nothing that recent studies have argued that part of detected SOAi tracers like 3-MeTHF-3,4-diols, C5-alkene triols and 2-methyltetrols are decomposition products of low-volatile IEPOX accretion products (like oligomers of IEPOX), which could occur during thermal desorption and/or derivatization heating processes64,65. Thus the tracers in the current study were possibly somewhat overestimated.
OC and EC in the PM2.5 samples were analyzed using DRI Model 2001 Carbon analyzer following the Interagency Monitoring of Protected Visual Environments (IMPROVE) thermal/optical reflectance (TOR) protocol. Water soluble inorganic ions including SO42−, NO3−, NH4+, Cl−, Ca2+, K+, and Mg2+ in samples were analyzed using ion chromatography. Detailed methods for OC, EC, and ions determination are provided in the supplementary material.
Estimations of aerosol pH and water content
ISORROPIA-II66,67, a thermodynamic model, was used to estimate aerosol pH and water content, because a direct measurement of aerosol acidity (i.e., pH) is not possible at present. Following by the assumptions of Weber et al.49, the PM2.5 aerosol at the Gucheng rural site was considered as internally mixed and in a single aqueous phase that contained the inorganic species, and no compositional dependence on particle size. In applying ISORROPIA-II, “forward” mode calculation, in which inputs to the model include temperature (T), relative humidity (RH), and the total (gas+aerosol) concentrations of aerosol precursors in the air parcel, would be more suitable to the aerosol pH and water content estimation, although the concentration of NH3, HNO3, and HCl in the gas phase are unavailable in the current study50. Therefore, the pH results given by the current work would be systematically underestimated by approximately one unit49,50.
References
Hallquist, M. et al. The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos. Chem. Phys. 9, 5155–5236 (2009).
Turpin, B. J. & Huntzicker, J. J. Identification of secondary organic aerosol episodes and quantitation of primary and secondary organic aerosol concentrations during SCAQS. Atmos. Environ. 29, 3527–3544 (1995).
Kanakidou, M. et al. Organic aerosol and global climate modelling: a review. Atmos. Chem. Phys. 5, 1053–1123 (2005).
Rollins, A. W. et al. Evidence for NOx Control over Nighttime SOA Formation. Science 337, 1210–1212, https://doi.org/10.1126/science.1221520 (2012).
Huang, R.-J. et al. High secondary aerosol contribution to particulate pollution during haze events in China. Nature 514, 218–222, https://doi.org/10.1038/nature13774 (2014).
Claeys, M. et al. Formation of secondary organic aerosols through photooxidation of isoprene. Science 303, 1173–1176 (2004).
Guenther, A. et al. A global model of natural volatile organic compound emissions. J. Geophys. Res.-Atmos. 100, 8873–8892, https://doi.org/10.1029/94jd02950 (1995).
Guenther, A. et al. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature). Atmos. Chem. Phys. 6, 3181–3210 (2006).
Piccot, S. D., Watson, J. J. & Jones, J. W. A global inventory of volatile organic compound emissions from anthropogenic sources. J. Geophys. Res.-Atmos. 97, 9897–9912 (1992).
Heald, C. L. et al. Predicted change in global secondary organic aerosol concentrations in response to future climate, emissions, and land use change. J. Geophys. Res.-Atmos. 113, https://doi.org/10.1029/2007jd009092 (2008).
Claeys, M. et al. Formation of secondary organic aerosols from isoprene and its gas-phase oxidation products through reaction with hydrogen peroxide. Atmos. Environ. 38, 4093–4098, https://doi.org/10.1016/j.atmosenv.2004.06.001 (2004).
Surratt, J. D. et al. Reactive intermediates revealed in secondary organic aerosol formation from isoprene. Proceedings of the National Academy of Sciences of the United States of America 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107 (2010).
Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Jaoui, M. & Edney, E. O. The formation of secondary organic aerosol from the isoprene plus OH reaction in the absence of NO(x). Atmos. Chem. Phys. 9, 6541–6558 (2009).
Lin, Y. H. et al. Isoprene Epoxydiols as Precursors to Secondary Organic Aerosol Formation: Acid-Catalyzed Reactive Uptake Studies with Authentic Compounds. Environ. Sci. Technol. 46, 250–258, https://doi.org/10.1021/es202554c (2012).
Paulot, F. et al. Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene. Science 325, 730–733, https://doi.org/10.1126/science.1172910 (2009).
Lin, Y.-H. et al. Epoxide as a precursor to secondary organic aerosol formation from isoprene photooxidation in the presence of nitrogen oxides. Proceedings of the National Academy of Sciences 110, 6718–6723, https://doi.org/10.1073/pnas.1221150110 (2013).
Nguyen, T. B. et al. Mechanism of the hydroxyl radical oxidation of methacryloyl peroxynitrate (MPAN) and its pathway toward secondary organic aerosol formation in the atmosphere. Physical Chemistry Chemical Physics 17, 17914–17926, https://doi.org/10.1039/c5cp02001h (2015).
Riva, M., Budisulistiorini, S. H., Zhang, Z., Gold, A. & Surratt, J. D. Chemical characterization of secondary organic aerosol constituents from isoprene ozonolysis in the presence of acidic aerosol. Atmos. Environ. 130, 5–13, https://doi.org/10.1016/j.atmosenv.2015.06.027 (2016).
Inomata, S. et al. Analysis of secondary organic aerosols from ozonolysis of isoprene by proton transfer reaction mass spectrometry. Atmos. Environ. 97, 397–405, https://doi.org/10.1016/j.atmosenv.2014.03.045 (2014).
Nguyen, T. B., Roach, P. J., Laskin, J., Laskin, A. & Nizkorodov, S. A. Effect of humidity on the composition of isoprene photooxidation secondary organic aerosol. Atmos. Chem. Phys. 11, 6931–6944, https://doi.org/10.5194/acp-11-6931-2011 (2011).
Zhang, H., Surratt, J. D., Lin, Y. H., Bapat, J. & Kamens, R. M. Effect of relative humidity on SOA formation from isoprene/NO photooxidation: enhancement of 2-methylglyceric acid and its corresponding oligoesters under dry conditions. Atmos. Chem. Phys. 11, 6411–6424, https://doi.org/10.5194/acp-11-6411-2011 (2011).
Surratt, J. D. et al. Effect of acidity on secondary organic aerosol formation from isoprene. Environ. Sci. Technol. 41, 5363–5369, https://doi.org/10.1021/es0704176 (2007).
Szmigielski, R. et al. The acid effect in the formation of 2-methyltetrols from the photooxidation of isoprene in the presence of NO(x). Atmospheric Research 98, 183–189, https://doi.org/10.1016/j.atmosres.2010.02.012 (2010).
Ding, X., Wang, X.-M. & Zheng, M. The influence of temperature and aerosol acidity on biogenic secondary organic aerosol tracers: Observations at a rural site in the central Pearl River Delta region, South China. Atmos. Environ. 45, 1303–1311 (2011).
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C. & Seinfeld, J. H. Secondary organic aerosol formation from isoprene photooxidation. Environ. Sci. Technol. 40, 1869–1877, https://doi.org/10.1021/es0524301 (2006).
Edney, E. O. et al. Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NO(X)/SO(2)/air mixtures and their detection in ambient PM(2.5) samples collected in the eastern United States. Atmos. Environ. 39, 5281–5289, https://doi.org/10.1016/j.atmosenv.2005.05.031 (2005).
Lin, Y. H., Knipping, E. M., Edgerton, E. S., Shaw, S. L. & Surratt, J. D. Investigating the influences of SO2 and NH3 levels on isoprene-derived secondary organic aerosol formation using conditional sampling approaches. Atmos. Chem. Phys. 13, 8457–8470, https://doi.org/10.5194/acp-13-8457-2013 (2013).
Zhang, Q. et al. Asian emissions in 2006 for the NASA INTEX-B mission. Atmos. Chem. Phys. 9, 5131–5153, https://doi.org/10.5194/acp-9-5131-2009 (2009).
Wang, G. H. et al. Persistent sulfate formation from London Fog to Chinese haze. Proceedings of the National Academy of Sciences of the United States of America 113, 13630–13635, https://doi.org/10.1073/pnas.1616540113 (2016).
Wang, G. H. et al. Organic Molecular Compositions and Size Distributions of Chinese Summer and Autumn Aerosols from Nanjing: Characteristic Haze Event Caused by Wheat Straw Burning. Environ. Sci. Technol. 43, 6493–6499, https://doi.org/10.1021/es803086g (2009).
Wang, J. et al. Concentrations and stable carbon isotope compositions of oxalic acid and related SOA in Beijing before, during, and after the 2014 APEC. Atmos. Chem. Phys. 17, 981–992, https://doi.org/10.5194/acp-17-981-2017 (2017).
Ding, X. et al. Spatial and seasonal variations of isoprene secondary organic aerosol in China: Significant impact of biomass burning during winter. Scientific reports 6, 20411, https://doi.org/10.1038/srep20411 (2016).
Lin, W., Xu, X., Ge, B. & Zhang, X. Characteristics of gaseous pollutants at Gucheng, a rural site southwest of Beijing. Journal of Geophysical Research 114, https://doi.org/10.1029/2008JD010339 (2009).
Zhao, X. J. et al. Analysis of a winter regional haze event and its formation mechanism in the North China Plain. Atmos. Chem. Phys. 13, 5685–5696, https://doi.org/10.5194/acp-13-5685-2013 (2013).
Fu, P. Q. et al. Organic molecular compositions and temporal variations of summertime mountain aerosols over Mt. Tai, North China Plain. J. Geophys. Res.-Atmos. 113, D19107, https://doi.org/10.1029/2008jd009900 (2008).
Wang, W. T. et al. Atmospheric polycyclic aromatic hydrocarbon concentrations and gas/particle partitioning at background, rural village and urban sites in the North China Plain. Atmospheric Research 99, 197–206, https://doi.org/10.1016/j.atmosres.2010.10.002 (2011).
Fu, P. Q., Kawamura, K., Kanaya, Y. & Wang, Z. F. Contributions of biogenic volatile organic compounds to the formation of secondary organic aerosols over Mt Tai, Central East China. Atmos. Environ. 44, 4817–4826, https://doi.org/10.1016/j.atmosenv.2010.08.040 (2010).
Fu, P. Q. et al. Diurnal variations of organic molecular tracers and stable carbon isotopic composition in atmospheric aerosols over Mt. Tai in the North China Plain: an influence of biomass burning. Atmos. Chem. Phys. 12, 8359–8375, https://doi.org/10.5194/acp-12-8359-2012 (2012).
Guo, S. et al. Primary Sources and Secondary Formation of Organic Aerosols in Beijing, China. Environ. Sci. Technol. 46, 9846–9853, https://doi.org/10.1021/es2042564 (2012).
Wang, W. et al. Characterization of oxygenated derivatives of isoprene related to 2-methyltetrols in Amazonian aerosols using trimethylsilylation and gas chromatography/ion trap mass spectrometry. Rapid communications in mass spectrometry: RCM 19, 1343–1351, https://doi.org/10.1002/rcm.1940 (2005).
Surratt, J. D. et al. Chemical composition of secondary organic aerosol formed from the photooxidation of isoprene. J. Phys. Chem. A 110, 9665–9690, https://doi.org/10.1021/jp061734m (2006).
Jason, D. Surratt et al. Chemical Composition of Secondary Organic Aerosol Formed from the Photooxidation of Isoprene. J. Phys. Chem. A 110, 9665 (2006).
Budisulistiorini, S. H. et al. Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site. Atmos. Chem. Phys. 15, 8871–8888, https://doi.org/10.5194/acp-15-8871-2015 (2015).
Rattanavaraha, W. et al. Assessing the impact of anthropogenic pollution on isoprene-derived secondary organic aerosol formation in PM2.5 collected from the Birmingham, Alabama, ground site during the 2013 Southern Oxidant and Aerosol Study. Atmos. Chem. Phys. 16, 4897–4914, https://doi.org/10.5194/acp-16-4897-2016 (2016).
Kourtchev, I., Warnke, J., Maenhaut, W., Hoffmann, T. & Claeys, M. Polar organic marker compounds in PM2.5 aerosol from a mixed forest site in western Germany. Chemosphere 73, 1308–1314 (2008).
Zhang, Y. et al. Limited formation of isoprene epoxydiols-derived secondary organic aerosol under NOx-rich environments in Eastern China. Geophysical Research Letters, https://doi.org/10.1002/2016gl072368 (2017).
Offenberg, J. H., Lewandowski, M., Edney, E. O., Kleindienst, T. E. & Jaoui, M. Influence of Aerosol Acidity on the Formation of Secondary Organic Aerosol from Biogenic Precursor Hydrocarbons. Environ. Sci. Technol. 43, 7742–7747, https://doi.org/10.1021/es901538e (2009).
Xu, L. et al. Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States. Proceedings of the National Academy of Sciences of the United States of America 112, 37–42, https://doi.org/10.1073/pnas.1417609112 (2015).
Weber, R. J., Guo, H., Russell, A. G. & Nenes, A. High aerosol acidity despite declining atmospheric sulfate concentrations over the past 15 years. Nature Geoscience 9, 282–285, https://doi.org/10.1038/ngeo2665 (2016).
Hennigan, C. J., Izumi, J., Sullivan, A. P., Weber, R. J. & Nenes, A. A critical evaluation of proxy methods used to estimate the acidity of atmospheric particles. Atmos. Chem. Phys. 15, 2775–2790, https://doi.org/10.5194/acp-15-2775-2015 (2015).
Liu, J. et al. Observational Evidence for Pollution influenced Selective Uptake Contributing to Biogenic Secondary Organic Aerosols in the Southeastern US. Geophysical Research Letters (2017).
Gaston, C. J. et al. Reactive uptake of an isoprene-derived epoxydiol to submicron aerosol particles. Environ Sci Technol 48, 11178–11186, https://doi.org/10.1021/es5034266 (2014).
Riedel, T. P. et al. Heterogeneous Reactions of Isoprene-Derived Epoxides: Reaction Probabilities and Molar Secondary Organic Aerosol Yield Estimates. Environmental Science & Technology Letters 2, 38–42, https://doi.org/10.1021/ez500406f (2015).
Budisulistiorini, S. H. et al. Simulating Aqueous-Phase Isoprene-Epoxydiol (IEPOX) Secondary Organic Aerosol Production During the 2013 Southern Oxidant and Aerosol Study (SOAS). Environ. Sci. Technol. 51, 5026–5034, https://doi.org/10.1021/acs.est.6b05750 (2017).
Li, J. J., Wang, G. H., Cao, J. J., Wang, X. M. & Zhang, R. J. Observation of biogenic secondary organic aerosols in the atmosphere of a mountain site in central China: temperature and relative humidity effects. Atmos. Chem. Phys. 13, 11535–11549, https://doi.org/10.5194/acp-13-11535-2013 (2013).
Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Jaoui, M. & Edney, E. O. Ozone-isoprene reaction: Re-examination of the formation of secondary organic aerosol. Geophysical Research Letters 34, https://doi.org/10.1029/2006gl027485 (2007).
Nguyen, T. B. et al. High-resolution mass spectrometry analysis of secondary organic aerosol generated by ozonolysis of isoprene. Atmos. Environ. 44, 1032–1042, https://doi.org/10.1016/j.atmosenv.2009.12.019 (2010).
Akagi, S. K. et al. Emission factors for open and domestic biomass burning for use in atmospheric models. Atmos. Chem. Phys. 11, 4039–4072, https://doi.org/10.5194/acp-11-4039-2011 (2011).
Andreae, M. O. & Merlet, P. Emission of trace gases and aerosols from biomass burning. Global Biogeochemical Cycles 15, 955–966, https://doi.org/10.1029/2000gb001382 (2001).
Zhu, Y. et al. Characteristics of ambient volatile organic compounds and the influence of biomass burning at a rural site in Northern China during summer 2013. Atmos. Environ. 124, 156–165, https://doi.org/10.1016/j.atmosenv.2015.08.097 (2016).
Gilardoni, S. et al. Direct observation of aqueous secondary organic aerosol from biomass-burning emissions. Proceedings of the National Academy of Sciences of the United States of America 113, 10013–10018, https://doi.org/10.1073/pnas.1602212113 (2016).
Wang, G. H., Kawamura, K., Lee, S., Ho, K. F. & Cao, J. J. Molecular, seasonal, and spatial distributions of organic aerosols from fourteen Chinese cities. Environ. Sci. Technol. 40, 4619–4625, https://doi.org/10.1021/es060291x (2006).
Wang, G. H. et al. Aircraft measurement of organic aerosols over China. Environ. Sci. Technol. 41, 3115–3120, https://doi.org/10.1021/es062601h (2007).
Lopez-Hilfiker, F. D. et al. Molecular Composition and Volatility of Organic Aerosol in the Southeastern U.S.: Implications for IEPOX Derived SOA. Environ Sci Technol 50, 2200–2209, https://doi.org/10.1021/acs.est.5b04769 (2016).
Hu, W. W. et al. Characterization of a real-time tracer for isoprene epoxydiols-derived secondary organic aerosol (IEPOX-SOA) from aerosol mass spectrometer measurements. Atmos. Chem. Phys. 15, 11807–11833, https://doi.org/10.5194/acp-15-11807-2015 (2015).
Fountoukis, C. & Nenes, A. ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+-Ca2+-Mg2+-Nh(4)(+)-Na+-SO42–NO3–Cl–H2O aerosols. Atmos. Chem. Phys. 7, 4639–4659 (2007).
Nenes, A., Pandis, S. N. & Pilinis, C. ISORROPIA: A new thermodynamic equilibrium model for multiphase multicomponent inorganic aerosols. Aquatic Geochemistry 4, 123–152, https://doi.org/10.1023/a:1009604003981 (1998).
Stein, A. F. et al. NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System. Bulletin of the American Meteorological Society 96, 2059–2077, https://doi.org/10.1175/bams-d-14-00110.1 (2015).
Rolph, G., Stein, A. & Stunder, B. Real-time Environmental Applications and Display sYstem: READY. Environmental Modelling & Software 95, 210–228, https://doi.org/10.1016/j.envsoft.2017.06.025 (2017).
Acknowledgements
This work was financially supported by the China National Natural Science Founds for Distinguished Young Scholars (Grants 41325014), the National Key R&D Plan (Quantitative Relationship and Regulation Principle between Regional Oxidation Capacity of Atmospheric and Air Quality) (No. 2017YFC0210000), the program from National Nature Science Foundation of China (No. 41773117) (No. 41405122, 91543116) and the West Light Foundation of Chinese Academy of Sciences. The authors gratefully acknowledge the use of fire spots data products from the Land, Atmosphere Near real-time Capability for EOS (LANCE) system operated by the NASA/GSFC/Earth Science Data and Information System (ESDIS) with funding provided by NASA/HQ (https://firms.modaps.eosdis.nasa.gov/firemap/), and the NOAA Air Resources Laboratory (ARL) for the provision of the HYSPLIT transport and dispersion model and/or READY website (http://www.ready.noaa.gov) used in this publication.
Author information
Authors and Affiliations
Contributions
G.H.W. designed the experiment. G.H.W., T.Z. and L.M.Z. arranged the sample collection. J.J.L., G.H.W., C.W., C.C., Y.Q.R., J.Y.W. and J.L. collected and analyzed the samples. J.J.L. and G.H.W. performed the data interpretation. G.H.W. and J.J.L. wrote the paper. T.Z. and J.J.C. contributed to manuscript revision.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, J., Wang, G., Wu, C. et al. Characterization of isoprene-derived secondary organic aerosols at a rural site in North China Plain with implications for anthropogenic pollution effects. Sci Rep 8, 535 (2018). https://doi.org/10.1038/s41598-017-18983-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18983-7
This article is cited by
-
Modeling Secondary Organic Aerosols in China: State of the Art and Perspectives
Current Pollution Reports (2023)
-
The effect of earth-air passive exchange on the formation of haze patterns
Environmental Earth Sciences (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
