
Abstract
Oil palm (Elaeis guineensis Jacq.) is the leading oil-producing crops and the most important edible oil resource worldwide. DNA markers and genetic linkage maps are essential resources for marker-assisted selection to accelerate genetic improvement. We conducted RAD-seq on an Illumina NextSeq500 to discover genome-wide SNPs, and used the SNPs to construct a linkage map for an oil palm (Tenera) population derived from a cross between a Deli Dura and an AVROS Pisifera. The RAD-seq produced 1,076 million single-end reads across the breeding population containing 155 trees. Mining this dataset detected 510,251 loci. After filtering out loci with low accuracy and more than 20% missing data, 11,394 SNPs were retained. Using these SNPs, in combination with 188 anchor SNPs and 123 microsatellites, we constructed a linkage map containing 10,023 markers covering 16 chromosomes. The map length is 2,938.2 cM with an average marker space of 0.29 cM. The large number of SNPs will supply ample choices of DNA markers in analysing the genetic diversity, population structure and evolution of oil palm. This high-density linkage map will contribute to mapping quantitative trait loci (QTL) for important traits, thus accelerating oil palm genetic improvement.
Similar content being viewed by others

Introduction
Oil palm (Elaeis guineensis Jacq) is a leading oil yielding crop in the world, mainly cultivated in the equatorial tropics of Africa, Southeast Asia and South America. Palm oil currently dominates the global vegetable oil economy, contributing to 33% of the total world oil production and 45% of edible oil worldwide1. However, one of the major constraints of oil palm production is slow improvement of oil yield per year2. To date, although conventional breeding in combination with improved field management has increased crude palm oil yield from 2.6 tons/ha to 4.0 Tons/ha during the past sixty years, there is still a big yield gap between that and the estimated theoretical potential of 18.2 tons/ha3.
To accelerate genetic improvement and to study biological questions in ecology and evolution, polymorphic DNA markers are required. Over the past two decades, DNA markers have been widely used for study of genetics, ecology, evolution and conservation in both model and non-model biology4. One important type of DNA markers is microsatellites. They are highly polymorphic and occur over ten-thousand locations in a species’ genome5. However, microsatellites exist preferentially in heterochromatic regions, and it is difficult to cover the whole genome evenly. In contrast, single nucleotide polymorphisms (SNPs) are widespread nucleotide variations in genomes, the most abundant type of DNA marker, and easily detected at high-throughput in plant and animal genomes4. By taking advantage of the recently developed reference genome sequences for plants and animals, it is easy to develop genome-wide DNA markers with gene information resources for study of genetics and evolution6. Furthermore, gene-based SNP markers could themselves be causative SNPs for traits, and have been used for developing transcript maps, QTL analysis, association mapping, and synteny analysis in crops7. Nevertheless, few oil palm SNPs have been applied in genetic and genomic research8,9,10.
Genome-wide DNA markers are required to construct high-resolution linkage maps. High-density linkage maps are essential for mapping QTL for marker-assisted selection (MAS) to speed up genetic improvement11,12,13,14, for the discovery of genomic regions responsible for both commercial and adaptive traits in evolutionary biology15, the study of genome evolution16 and the chromosomal structure variation17, as well as for genome assembly18,19. For oil palm, the first linkage map was constructed based on restriction fragment length polymorphism (RFLP) markers20. The other linkage maps were made using AFLPs (Amplified fragment length polymorphisms), microsatellites and SNPs markers8,9,21,22,23. However, these maps were unsaturated and most contained only hundreds of markers, largely consisting of microsatellite markers and a few SNPs, which are not sufficient to accurately map QTL for important traits. A high-density genetic map is critically important to accurately map QTL for important traits for MAS, and ultimately to accelerate genetic improvement.
By combining the power of next generation sequencing (NGS) with reduced representation, the discovery, validation and assessment of genome-wide markers in studies have undergone a revolutionary transition over the past ten years with the advent of low-cost and high-throughput restriction-site associated DNA sequencing (RAD-seq) technology24. In comparison to traditional marker discovery, RAD-seq is less complicated for constructing highly multiplexed, reduced representation libraries even in species with large genomes25. It can simultaneously discover and genotype many (e.g. >2000) SNPs within a few days. This technique has successfully been applied to discover thousands of markers in plants such as wheat and barley26, sorghum27, rice28, switchgrass29, sesame30, and in insects and fish31. While more than ten genetic linkage maps are available for oil palm, only two linkage maps was based on around 1000 SNPs9,32.
The aim of our study was to discover many genome-wise SNPs and to construct an ultrahigh-density SNP-based linkage map for the oil palm. These identified genome-wide SNPs will facilitate studies on oil palm ecology and evolution, whereas the ultrahigh-density linkage map will be useful in discovering QTL for important traits for marker-assisted selection to accelerate genetic improvement.
Results
Microsatellite marker analysis
A total of 123 out of 300 microsatellite markers selected were informative between the two parental palms, a Deli Dura and an AVROS Pisifera. Both parental trees were used to produce F1 Tenera for production in Wilmar International Plantation. All the markers were used to genotype 153 F1 palms from the F1 production population. Finally, 123 microsatellite markers were mapped into the LGs together with the SNPs. The sequences of the microsatellite markers mapped are listed in Additional file Table S1.
SNP genotyping using micro-chip assay
A set of 288 SNP markers on 16 chromosomes were used to genotype 153 palms and two parental palms by using the 96.96 Dynamic Array Chip (Fluidigm, Singapore), then 188 SNPs were mapped into the linkage groups (LGs) together with other markers in the palm population. The sequence and alleles of the SNP markers mapped are listed in Additional Table S1. Among the 188 SNPs, 39 SNPs were developed from genes associated with lipid synthesis based on the genome annotation of oil palm33.
SNP discovery and genotyping using RAD-seq
Four RAD-seq libraries were constructed using 96 barcodes and the PstI-HF restriction endonuclease. A total of 1,076 million raw reads were obtained from the sequencing of 155 trees. After filtering out low quality and ambiguous barcodes, there remained 1,050 million reads (97.57% of total raw reads). The retained reads of the two parental palms were 11.73 and 8.89 million reads, respectively (Table 1). From the mapping population, 8 out of 153 progeny individuals were discarded from further analysis due to low coverage of sequencing reads. The average number of raw reads across all retained 145 progeny was 7.10 million.
Using the sequence data of the two parental samples, a catalogue containing 510,251 loci was built for SNP discovery. After removing the loci with >20% missing data across progeny, a total of 11,394 informative SNPs were genotyped and used for linkage map construction. Additional Table S1 lists, for each marker in the map, its alleles, flanking sequence, name of contig containing it, and its physical position in the contig.
Construction of a high-density linkage map
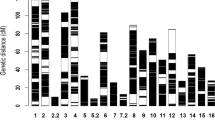
The 11,394 SNPs identified from RAD-seq, with another 188 SNPs from micro-chip array and 123 microsatellite markers, were used to construct the linkage map. Of these markers, 1,682 SNPs either could not be assigned to LGs or were assigned to small groups (marker number less than 50), or had segregation distortion (P < 0.001), were excluded from further analysis. The retained 10,023 markers were assigned to 16 LGs (Chromosomes) (Fig. 1). The linkage map contained 10,023 markers (9,900 SNPs and 123 microsatellites). The map covered a total length of 2,938.2 cM and had an average marker density of 0.29 cM. The lengths of LGs ranged from 53.47 cM for chromosome (Chr) 15 to 416.78 cM for Chr 1, with an average of 183.64 cM. The marker intervals ranged from 0.17 cM on Chr 10 to 0.46 cM on Chr 13, with an average of 0.29 cM. Marker spaces in all LGs are smaller than 20 cM, except in Chr 2 and Chr 13, with 30.13 cM and 33.39 cM, respectively. A summary of the markers, marker densities and genetic distances for each LG, as well as its corresponding chromosome in the draft genome1 are shown in Table 2 and Additional Table S1.

A ultrahigh-density linkage map of African oil palm (Elaeis guineensis) with 10,023 markers (i.e. 9900 SNPs markers and 124 microsatellites, see details about the mapped markers in Additional Table S1). The bar on left side represents the map length in cM, whereas the horizontal bars in each linkage group (chromosome) are positions of markers mapped.
Using the reference genome annotation resource1, we searched the genes associated with each SNP. Then 5,727 SNPs were annotated with gene information, with 3,975 SNPs located in intergenic regions. The number of SNPs with gene information in each of the 16 chromosomes ranged from 54 to 829 (Table 2 and Additional Table S1).
Discussion
After the RAD-seq approach was developed34,35, it was initially adopted in a population genomic study36. With the continuously decreasing cost of next-generation sequencing (NGS), RAD-seq has become a powerful approach for many research problems, from discovering thousands of SNPs for large-scale population genotyping purposes, to constructing the dense linkage maps for QTL mapping, scaffolding of newly sequenced genomes, and addressing a wide range of questions in molecular ecology and evolutionary biology37. Usually, ddRAD-seq with PstI-MspI is used for identification of SNPs in plants and animals18,38,39. The first SNP discovery in oil palm using ddRAD-seq with PstI-MspI was conducted with 3417 informative SNPs9. In a later study32, 2139 informative SNPs were identified using ddRAD-seq with PstI-MspI in an oil palm breeding population, and a high-density SNP genotyping array was developed40. In this study, we discovered more than 10,000 informative SNPs using a different approach: single digest RAD-seq with the PstI35, modified for a highly heterozygous breeding oil palm population. These indicate that both approaches of RAD-seq could facilitate SNP discovery and improvement of genomic resource. The significant difference in number of SNPs discovered between the two RAD-seq approaches could be caused by restriction enzyme selection, RAD-seq library preparation methods, coverage of sequences reads, as well as the heterozygosity of samples being used in the studies. The previous study showed that variance in read depth among individuals could be minimized, but the expected frequency of restriction cut was decided by a particular restriction enzyme34, hence ddRAD-seq with PstI-MspI could result in a lower digest efficiency of endonuclease and hence less common SNPs in the highly heterozygous oil palm samples. The previous study also indicated that the loci discovered across the genome showed the interaction between choice of restriction enzymes and the number of loci recovered from different parts of the genome41. Here we provide an evidence that the complexity reduction involving the restriction enzyme PstI-HF was adapted in order to avoid and lower the occurrence of repetitive regions of the high heterozygous oil palm genome. That, in combination with the 300–600 bp size selection for RAD library construction, the library single-end Illumina sequencing by Illumina NextSeq500 in our laboratory, the high throughput reads (150 bp per read) at sufficient depth of coverage which can provide sufficient contigs containing SNPs (Table 1), and reads sequenced with a mean coverage depth of 7~10× which should allow accurately genotyping at most sites across most individuals, means that a simple, highly efficient and lower cost RAD-seq platform for heterozygous oil palm samples has been constructed in this study. The entire process from DNA isolation, library preparation to sequence using Illumina NextSeq500, and SNP calling through sstacks pipeline, was relatively simple and fast. Furthermore, the overall cost of RAD-seq was economically efficient. Given the benefit of using this platform in our studies, it may be expected that utilization of this technology will become widely adopted in oil palm and other highly heterozygous samples for SNP discovery, genetic and genomic studies, and this approach has improved the tools developed, alleviating the marker limitation that researchers previously faced for studies of high heterozygous samples.
With the RAD-seq, we discovered over 10 K high-quality and informative SNP markers for oil palm. Among them, 5,727 SNPs are located in genes. These over 10 K SNPs will be useful in studies on ecology, evolution and linkage and QTL mapping for economically important traits to accelerate genetic improvement in oil palm. The genome-wide set of over 5 K SNP markers within genes could facilitate to identity causative SNPs within QTL for important traits, and can be useful in synteny analysis to understand more about genome evolution in plants7.
Based on SNPs discovered across the whole oil palm genome, we generated a genome-wide high-density linkage map containing 10,023 markers, distributed through all 16 chromosomes. The length is 2938.2 cM and the average marker density is 0.29 cM. Compared with previous studies, marker numbers in linkage maps have continued to increase, from 252 to 944 microsatellite-based markers8,21,22,23, to 1085 SNP-based markers9, and to the SNP-based ultra-high-density linkage map in this study. Also, in terms of marker density, our map is among the highest published to date, compared to the previous marker spacings of 1.26 cM9 and 1.4 cM8. The high-density linkage map can enable QTL mapping for the gaining of insights into the genome-wide genetic architecture of economic traits, genomic evolutionary studies, genome assembly, revealing the patterns of chromosomal evolution, and linkage disequilibria assessing4. Such maps are frequently constructed using microsatellite markers, but microsatellite markers are not as abundant or easily scored as SNPs4. However, we noticed that in Chr 2 and 13, in a few positions, marker spaces were bigger than 20 cM. More markers should be mapped in these positions to reduce marker interval for genomic studies. Improvement of the platform of RAD-seq with high efficiency library preparation and bioinformatics procedures used to identify SNPs, as well as the use of large populations for oil palm, could be a feasible solution for this challenge.
In conclusion, we identified many SNPs using RAD-seq, supplying sufficient DNA markers for studying ecology and evolution of oil palm. Then, we constructed an ultra-high-density linkage map with 10,023 markers for African oil palm. To the best of our knowledge, this is the densest linkage map in oil palm. The linkage map will facilitate mapping QTL for economically important traits for MAS to accelerate genetic improvement in oil palm.
Materials and Methods
The palm population used for linkage mapping
A F1 population was generated by crossing a Deli Dura (mother palm) and an AVROS Pisifera (father palm) in Wilmar International Plantation. Both parental trees were used to produce Tenera trees for production in Wilmar International Plantation. A total of 153 F1 progenies (Tenera type) were planted into a plantation field in Indonesia in 2006. These F1 trees were managed under the same conditions following the standard protocol of Wilmar Plantation in Indonesia (http://www.wilmar-international.com/our-business/tropical-oils/plantations/).
DNA extraction from palm leaves
DNA was isolated from young leaf samples using DNAeasy Plant Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instruction. RNA was removed by RNAse digestion during lysis, and cell debris, precipitated proteins, and polysaccharides were removed by centrifugation through a QIAshredder spin column. Then, the lysate was loaded into the DNEasy Plant Mini spin column, in which DNA selectively binds to the silica membrane. Finally, the remaining contaminants and enzyme inhibitors were removed in one or two efficient wash steps and pure DNA was then eluted in 1xTE buffer. The quality of DNA was checked on 1.0% agarose gels. DNA quantification was conducted using Nanodrop 2000 (Nanodrop, Wilmington, DE, USA). For RAD-seq library construction, the concentration of double-strand DNA was also measured using Quanti-Qubit® dsDNA HS Assay Kit (Thermo Fisher Scientific, Carlsbad, CA, USA) on a microplate reader of Tecan Infinite M200 (Tecan, Männedorf, Switzerland).
Genotyping of microsatellites
The microsatellites used in this study (see Additional Table S1) were developed by us22 using a method described previously42 Primers of each microsatellite were designed in the repeat-flanking regions using PrimerSelect (DNAstar, CA, USA). To genotype the microsartellites on the automatic DNA sequencer ABI3730xl (Applied Biosystems, Foster City, CA, USA) one primer for each marker was labelled with a fluorescent dye (i.e. either HEX or 6-FAM). The microsatellite markers were genotyped individually with PCR and electrophoresis for polymorphism and informativeness between the two parental trees. Each microsatellite marker was amplified using PCR, which contained 40 ng of genomic DNA, 2.0 units of DNA Taq-DNA-polymerase (Bio-Rad, Berkeley, CA, USA), 1× PCR buffer containing 1.5 mM MgCl2, 0.2 µM dNTPs and 50 nM of each primer. PCR was carried out on PTC100-PCR machines (Bio-Rad, Berkeley, CA, USA). The following PCR conditions were applied: three minutes of denaturation at 94 °C, 35 cycles of 30 s at 94 °C, 30 s at 53–58 °C and 45 s at 72 °C; and a final extension at 72 °C for 10 min. The PCR products were electrophoresed and analysed on an ABI3730x1 sequencer (Applied Biosystems, Foster City, CA, USA). The allele sizes and genotypes were analysed against the ROX-500 standard using GeneMapper 4.1 (Applied Biosystems, Foster City, CA, USA). The genotype data were exported for later linkage mapping.
Genotyping of SNPs using micro-chip based on allele-specific competitive PCR assay
From over 10,000 SNPs identified in the 17 re-sequenced palm genomes33, 288 SNPs were randomly selected from 16 chromosomes as anchor markers based on their genomic position from previously published genome1. The SNPs were genotyped using allele-specific competitive PCR assay according to our method43, which is a homogenous, fluorescence-based genotyping technology that is based on allele-specific oligo extension and fluorescence resonance energy transfer for signal generation. The SNP assay consisted of three parts. Firstly, the primer design: the SNP allele-specific forward primers and a common reverse primer were designed based on an approximately 120 bp long genomic sequence flanking each SNP site (http://bioinfo.biotec.or.th/WASP/). The two probes with fluoresce-nt labels (FAM and HEX, respectively) complement the tail sequence of the allele-specific forward primers. Secondly, loading the assay components: DNA sample (50 ng/µl), 2× Biotium Fast Probe qPCR Master Mix buffer, forward primer mix (0.25 µM), reverse primer (0.25 µM), probe primers mix (10 µM), ROX (50×), 20× sample loading reagent, and two probe primers were mixed together with the DNA sample and then loaded into the sample cell, and the two forward primers and the reverse primer were mixed together and loaded into primer cell in the 96.96 Dynamic ArrayTM IFC (Fluidigm, South San Francisco, CA, USA). Finally, the PCR reaction: The reaction was performed in a Biomark HD (Fluidigm, South San Francisco, CA, UAS) with following steps: 94 °C for 15 min; followed by 9 cycles of 94 °C for 20 s; and annealing at 65 °C (−1 °C per cycle) for 20 s, and 72 °C for 20 s; then 30 cycles of 94 °C for 10 s, 57 °C for 1 min and 72 °C for 20 s; and a final extension of 72 °C for 3 min. Then the SNP genotypes of the samples were analysed based on the two parental samples’ alleles.
Construction of RAD-seq libraries and sequencing
The RAD-seq libraries were constructed according to the published method35 with some modifications. A total of 153 trees of the breeding population and two parental trees were used for library construction. Briefly, 500 ng of genomic DNA from each individual was digested with high-fidelity PstI (New England Biolabs, Ipswich, MA, USA), and the digested fragments were then ligated to barcoded adapters. The ligation products with unique barcodes were pooled and sheared with Covaris M220 (Covaris, Woburn, MA USA) with a DNA fragment peak of 500 bp. The sheared DNA was purified with MinElute PCR purification kit (Qiagen, Hilden, Germany) and then size selected (300–600 bp) with Agencourt AMPure XP beads (Beckman coulter life sciences, Brea, CA, USA). Recovered DNA was blunt end repaired using Quick Blunting Kit (New England Biolabs, Ipswich, MA) and then A-overhangs were added with klenow fragment (New England Biolabs, Ipswich, MA, USA). Sequencing adaptor P2 was then ligated to the DNA fragments with T4 DNA ligase (New England Biolabs, Ipswich, MA, USA). Finally, the library was amplified using Phusion® High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA, USA) and the PCR products were purified using Agencourt AMPure XP beads. The recovered library was quantified with Illumina library quantification kit (Kapa Biosystems, Wilmington, MA, USA) in the MyiQ Thermal Cycler (Bio-Rad, Hercules, CA, USA) and sequenced for 1 × 150 bp reads on NextSeq500 (Illumina, San Diego, CA, USA).
Identifying of SNP from RAD-seq and genotyping
The raw sequencing reads were demultiplexed and cleaned using the program process_radtags in the software package Stacks v1.4234. Any uncalled base and low quality reads were removed. To minimize the sequencing errors, all the clean reads were trimmed to 130 bp. In detail, only the reads with a limit of >Q30 for Illumina sequencing were used for further analysis, the SE reads were then trimmed to 130 bp from the 3′ end and that of any uncalled base were removed. The reads with an average score within sliding window of <10 were also removed. The cleaned reads were aligned to the oil palm reference genome1 by the use of the program GSNAP with a maximum mismatch of 5 bp. Only the alignments of unique targets were used for stacks assembly using the program pstacks (−m, 3). The assembled stacks from the parents were used to build a catalogue of loci using the program cstacks in the Stacks package. The assembled stacks from each sample were then matched to the catalogue for SNP discovery using sstacks. Finally, the program genotypes was used to call SNPs across progeny. The genotyped SNPs were filtered with a cut-off of at least 10 × sequence depth and at most 20% missing data. Then the genes associated with SNP loci were searched against the oil palm genome annotation1. Markers heterozygous in any one parent or both parents were selected for further analysis. There are five different allelic patterns in all called markers: type nn × np (segregating 1:1) was homozygous in the male parent and heterozygous in the female parent; lm × ll (1:1) was heterozygous in the male parent and homozygous in the female parent; hk × hk (1:2:1) was heterozygous in both parents with two shared alleles; ef × eg (1:1:1:1) was heterozygous in both parents with two sex-specific alleles and one shared allele, and type ab × cd (1:1:1:1) was heterozygous in both parents with four different alleles.
Linkage map construction
Linkage analysis was performed for markers genotyped in F1 palms derived from crossing Dura and Pisifera. Polymorphic markers from microsatellites and anchor SNPs, and SNPs from RAD-seq, were used to construct the linkage map. The genetic linkage map was generated using software Lep-MAP244,45. Before linkage map analysis, the default dataTolerance value of 0.001 (χ2 test, P < 0.001) was used to filter out markers with segregation distortion. Then the remaining loci were grouped into linkage groups (LGs) using a LOD score threshold of 10. The order and positions of the markers within their respective LGs were determined according to the recombination probabilities between loci, with the sex-averaged option selected. And the genetic maps were drawn using the software MapChart V2.246. Finally, the LGs for the mapping population were numbered according to the published linkage map (Billotte et al.21) based on the anchor markers and genomic sequence1,21.
Data availability
The RAD-seq datasets supporting the conclusions of this article are available in the DDBJ database, accession number (BioProject Accession: PRJDB5817).
References
Singh, R. et al. Oil palm genome sequence reveals divergence of interfertile species in old and new worlds. Nature 500, 335–339 (2013).
Morcillo, F. et al. Improving palm oil quality through identification and mapping of the lipase gene causing oil deterioration. Nat. Commun. 4, 2160 (2013).
Barcelos, E. et al. Oil palm natural diversity and the potential for yield improvement. Front. Plant Sci. 6, 190 (2015).
Thomson, R. C., Wang, I. J. & Johnson, J. R. Genome-enabled development of DNA markers for ecology, evolution and conservation. Mol. Ecol. 19, 2184–2195 (2010).
Zane, L., Bargelloni, L. & Patarnello, T. Strategies for microsatellite isolation: a review. Mol. Ecol. 11, 1–16 (2002).
Backström, N., Fagerberg, S. & Ellegren, H. Genomics of natural bird populations: a gene‐based set of reference markers evenly spread across the avian genome. Mol. Ecol. 17, 964–980 (2008).
Galeano, C. H. et al. Gene-based single nucleotide polymorphism markers for genetic and association mapping in common bean. BMC Genet. 13, 48 (2012).
Ting, N.-C. et al. High density SNP and SSR-based genetic maps of two independent oil palm hybrids. BMC Genomics 15, 309 (2014).
Pootakham, W. et al. Genome-wide SNP discovery and identification of QTL associated with agronomic traits in oil palm using genotyping-by-sequencing (GBS). Genomics 105, 288–295 (2015).
Teh, C.-K. et al. Genome-wide association study identifies three key loci for high mesocarp oil content in perennial crop oil palm. Sci. Rep. 6 (2016).
Bodénès, C., Chancerel, E., Ehrenmann, F., Kremer, A. & Plomion, C. High-density linkage mapping and distribution of segregation distortion regions in the oak genome. DNA Res. 23, 115–124 (2016).
Lohithaswa, H. & Pandravada, A. S. Construction of high-density linkage map and identification of QTLs for resistance to sorghum downy mildew in maize (Zea mays L.). Mol. Breed. 37, 2 (2017).
Ma, Y. et al. Genome-wide SNP identification, linkage map construction and QTL mapping for seed mineral concentrations and contents in pea (Pisum sativum L.). BMC Plant Biol. 17, 43 (2017).
Xu, Y. & Crouch, J. H. Marker-assisted selection in plant breeding: from publications to practice. Crop Sci. 48, 391–407 (2008).
Tanksley, S. et al. High density molecular linkage maps of the tomato and potato genomes. Genetics 132, 1141–1160 (1992).
Nietlisbach, P. et al. A microsatellite‐based linkage map for song sparrows (Melospiza melodia). Mol. Ecol. Resour. 15, 1486–1496 (2015).
Palomar, G. et al. Comparative High-Density Linkage Mapping Reveals Conserved Genome Structure but Variation in Levels of Heterochiasmy and Location of Recombination Cold Spots in the Common Frog. G3 Genes Genom. Genet. 7, 637–645 (2016).
Wang, L. et al. Construction of high-resolution recombination maps in Asian seabass. BMC Genomics 18, 63 (2017).
Song, Q. et al. Construction of high resolution genetic linkage maps to improve the soybean genome sequence assembly Glyma1. 01. BMC Genomics 17, 33 (2016).
Mayes, S., Jack, P., Corley, R. & Marshall, D. Construction of a RFLP genetic linkage map for oil palm (Elaeis guineensis Jacq.). Genome 40, 116–122 (1997).
Billotte, N. et al. Microsatellite-based high density linkage map in oil palm (Elaeis guineensis Jacq.). Theor. Appl. Genet. 110, 754–765 (2005).
Lee, M. et al. A consensus linkage map of oil palm and a major QTL for stem height. Sci. Rep. 5, 8232 (2015).
Singh, R. et al. Mapping quantitative trait loci (QTLs) for fatty acid composition in an interspecific cross of oil palm. BMC Plant Biol. 9, 114 (2009).
Andrews, K. R., Good, J. M., Miller, M. R., Luikart, G. & Hohenlohe, P. A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nature Rev. Genet. 17, 81–92 (2016).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLOS ONE 6, e19379 (2011).
Poland, J. A., Brown, P. J., Sorrells, M. E. & Jannink, J.-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLOS ONE 7, e32253 (2012).
Morris, G. P. et al. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc. Natl. Acad. Sci. USA 110, 453–458 (2013).
Spindel, J. et al. Bridging the genotyping gap: using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor. Appl. Genet. 126, 2699–2716 (2013).
Lu, F. et al. Switchgrass genomic diversity, ploidy, and evolution: novel insights from a network-based SNP discovery protocol. PLOS Genet. 9, e1003215 (2013).
Uncu, A. O., Frary, A., Karlovsky, P. & Doganlar, S. High-throughput single nucleotide polymorphism (SNP) identification and mapping in the sesame (Sesamum indicum L.) genome with genotyping by sequencing (GBS)analysis. Mol. Breed. 36, 173 (2016).
Burford Reiskind, M. et al. Development of a universal double‐digest RAD sequencing approach for a group of nonmodel, ecologically and economically important insect and fish taxa. Mol. Ecol. Resour. 16, 1303–1314 (2016).
Bai, B. et al. Genome-wide identification of markers for selecting higher oil content in oil palm. BMC Plant Biol. 17, 93 (2017).
Jin, J. et al. Draft genome sequence of an elite Dura palm and whole-genome patterns of DNA variation in oil palm. DNA Res. 23, 527–533 (2016).
Catchen, J. M., Amores, A., Hohenlohe, P., Cresko, W. & Postlethwait, J. H. Stacks: building and genotyping loci de novo from short-read sequences. G3 Genes Genom. Genet. 1, 171–182 (2011).
Baird, N. A. et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLOS ONE 3, e3376 (2008).
Hohenlohe, P. A. et al. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLOS Genet. 6, e1000862 (2010).
Davey, J. W. & Blaxter, M. L. RADSeq: next-generation population genetics. Brief. Funct. Genomics. 9, 416–423 (2010).
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S. & Hoekstra, H. E. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLOS ONE 7, e37135 (2012).
Sonah, H., O’Donoughue, L., Cober, E., Rajcan, I. & Belzile, F. Identification of loci governing eight agronomic traits using a GBS‐GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol. J. 13, 211–221 (2015).
Qi, B. K. et al. Development and Validation of a High-Density SNP Genotyping Array for African Oil Palm. Molecular Plant 9, 1132 (2016).
DaCosta, J. M. & Sorenson, M. D. Amplification biases and consistent recovery of loci in a double-digest RAD-seq protocol. PLOS ONE 9, e106713 (2014).
Yue, G., Chen, F. & Orban, L. Rapid isolation and characterization of microsatellites from the genome of Asian arowana (Scleropages formosus, Osteoglossidae, Pisces). Mol. Ecol. 9, 1007–1009 (2000).
Wang, L. et al. Genome-wide discovery of gene-related SNPs in Barramundi Lates calcarifer. Conserv. Genet. Resour. 7, 605–608 (2015).
Rastas, P., Paulin, L., Hanski, I., Lehtonen, R. & Auvinen, P. Lep-MAP: fast and accurate linkage map construction for large SNP datasets. Bioinformatics 29, 3128–3134 (2013).
Rastas, P., Calboli, F. C., Guo, B., Shikano, T. & Merilä, J. Construction of ultradense linkage maps with Lep-MAP2: stickleback F2 recombinant crosses as an example. Genome Biol. Evol. 8, 78–93 (2016).
Voorrips, R. MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78 (2002).
Acknowledgements
We thank staff members of the R&D department of Wilmar International Plantation for technical support.
Author information
Authors and Affiliations
Contributions
N.H.C. initiated and coordinated the project “Genetic Improvement of Oil Palm”. G.H.Y. designed the experiment and supervised the lab work. B.B. performed SNP development, linkage map construction, and contributed to drafting the manuscript. L.W. assisted in RAD-seq data analysis and SNP calling. Y.J.Z. performed the sample preparation and DNA extraction, M.L. prepared DNA samples and genotyping for gene-based SNP and microsatellite markers, B.Q.Y. contributed to the comparative analysis and editing of the paper. Rahmadsyah, C.H.L., Y.A., S.P. and A.S. managed plants in the field and were involved in developing microsatellite markers. G.H.Y. finalized the manuscript, and all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors B.B., L.W. Y.J.Z., M.L., B.Q.Y., N.H.C. and G.H.Y. declare that they have no competing interests. The authors R.,Y.A., S.P. and A.S. are employees of Wilmar International and Wilmar International funded this study. Therefore, they have financial competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bai, B., Wang, L., Zhang, Y.J. et al. Developing genome-wide SNPs and constructing an ultrahigh-density linkage map in oil palm. Sci Rep 8, 691 (2018). https://doi.org/10.1038/s41598-017-18613-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18613-2
This article is cited by
-
Combination of GWAS and FST-based approaches identified loci associated with economic traits in sugarcane
Molecular Genetics and Genomics (2023)
-
Compatibility between snails and schistosomes: insights from new genetic resources, comparative genomics, and genetic mapping
Communications Biology (2022)
-
Genome properties of key oil palm (Elaeis guineensis Jacq.) breeding populations
Journal of Applied Genetics (2022)
-
Sequence-Related Amplified Polymorphism (SRAP) Markers Based Genetic Diversity and Population Structure Analysis of Oil Palm (Elaeis guineensis Jacq.)
Tropical Plant Biology (2021)
-
Molecular approaches for improving oil palm for oil
Molecular Breeding (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
