
Abstract
Peroxisome proliferator-activated receptor γ (PPARγ) has received significant attention as a key regulator of glucose and lipid homeostasis. In this study, we synthesized and tested a library of novel 5-benzylidene-thiazolidin-2,4-dione (BTZD) derivatives bearing a substituent on nitrogen of TZD nucleus (compounds 1a-1k, 2i-10i, 3a, 6a, and 8a-10a). Three compounds (1a, 1i, and 3a) exhibited selectivity towards PPARγ and were found to be weak to moderate partial agonists. Surface Plasmon Resonance (SPR) results demonstrated binding affinity of 1a, 1i and 3a towards PPARγ. Furthermore, docking experiments revealed that BTZDs interact with PPARγ through a distinct binding mode, forming primarily hydrophobic contacts with the ligand-binding pocket (LBD) without direct H-bonding interactions to key residues in H12 that are characteristic of full agonists. In addition, 1a, 1i and 3a significantly improved hyperglycemia and hyperlipidaemia in streptozotocin-nicotinamide (STZ-NA)-induced diabetic rats at a dose of 36 mg/kg/day administered orally for 15 days. Histopathological investigations revealed that microscopic architecture of pancreatic and hepatic cells improved in BTZDs-treated diabetic rats. These findings suggested that 1a, 1i and 3a are very promising pharmacological agents by selectively targeting PPARγ for further development in the clinical treatment of type 2 diabetes mellitus.
Similar content being viewed by others
Introduction
Type 2 diabetes mellitus (T2DM), formerly referred to as non-insulin dependent diabetes mellitus (NIDDM), has become a major public health issue that affects more than 285 million people worldwide1. It is estimated that by 2030 around 552 million people will have diabetes2. T2DM is a complex heterogeneous group of metabolic disorders resulting in high blood glucose due to impaired insulin secretion or insulin resistance. The major factors contributing to the development of abnormal insulin secretion or insulin resistance include genetic defects, aging, viral infection (Cytomegalovirus, Coxsackie B4 virus), environmental factors, sedentary lifestyle combined with high calorie intake3. T2DM leads to further long term complications such as neuropathy, nephropathy, cataracts and accelerated atherosclerosis leading to increased risk of myocardial infarction4,5,6. In fact, the prevention and control of associated complications are a serious and challenging therapeutic problem, as these factors represent the leading causes of mortality and morbidity for diabetic patients7.
Peroxisome Proliferator-Activated Receptors (PPARs) represent a group of transcription factors that, upon binding of a ligand, induce the expression of genes involved in the regulation of glucose homeostasis and lipid metabolism and for this reason they have been considered suitable targets for the treatment of metabolic disorders8. In humans, three different subtypes of these nuclear receptors have been identified: PPARα (NR1C1), PPARβ (also known as PPARδ) (NR1C2), and PPARγ (NR1C3)9. PPARα, that is the target of hypolipidemic drugs fibrates, is highly expressed in brown adipose tissue, liver, kidney, heart and skeletal muscle and activates a genetic program leading to β-oxidation of fatty acids10,11. PPARβ/δ is localized ubiquitously but its function remains as the least understood amongst PPAR subtypes although numerous studies have highlighted the physiological importance of PPARβ/δ in the regulation of fatty acid catabolism and energy homeostasis12,13. PPARγ is expressed in three different forms PPARγ1, PPARγ2 and PPARγ314. PPARγ1 is expressed in heart, kidney, pancreas, colon, spleen; PPARγ2 is expressed in adipose tissue and PPARγ3 is predominantly found in white adipose tissue, macrophages and large intestine15. PPARγ acts as a key factor in various metabolic processes and plays an important role in the regulation of insulin tissue sensitivity and in the management of glucose and lipid uptake and storage16. Being expressed in many components of the vascular and immune system, PPARγ also exerts anti-inflammatory and anti-atherosclerotic actions17,18,19. This nuclear receptor is the target of thiazolidinedione class of insulin sensitizing drugs, used in T2DM20. The antihyperglycemic properties of thiazolidinediones (TZDs) came into existence by description of ciglitazone in the early 1980s and were followed by a few more glitazones in subsequent years (Fig. S1)21,22,23,24. Due to the associated toxicity25,26,27, some of these drugs as well as other PPARγ full agonists have been abandoned during clinical trials or withdrawn from the market28. Nowadays, therefore, new drugs acting as partial agonists, even weak or moderate, or selective modulators of PPARγ (SPPARγM) are being developed with the goal of retaining the beneficial effects while reducing the adverse effects29,30,31,32,33,34. In the past, very few 5-benzylidene-thiazolidin-2,4-dione (BTZD) derivatives have been explored for their glucose lowering and selective PPARγ modulator activity35,36,37,38,39.
In search for less toxic derivatives of TZDs, in the present paper we synthesized a library of novel BTZD derivatives bearing a substituent on the nitrogen atom of TZD nucleus (compounds 1a-1k, 2i-10i, 3a, 6a, and 8a-10a) and tested them for their ability to bind and activate PPARγ by SPR experiments and transactivation assays, respectively. Among these derivatives, we identified three novel BTZD derivatives as PPARγ partial agonists (1a, 1i, and 3a). In addition, we demonstrated by means of docking experiments that BTZDs utilize a binding mode distinct from other reported PPARγ ligands, without making direct contact with any residues of helix 12. Moreover, we investigated the antidiabetic effects of 1a, 1i, and 3a in streptozotocin-nicotinamide (STZ-NA)-induced diabetic rats, which proved that BTZDs improved metabolic parameters and lipid profile. These compounds had also favourable effect to inhibit the histopathological changes of the pancreas and kidney in STZ-NA-induced diabetic rats. Finally, we studied the oral bioavailability of 1a, 1i, and 3a in terms of Caco-2 and MDCK-II permeability (Papp,A–B) and the P-gp efflux ratio (Papp,B–A/Papp,A–B). The in vitro intrinsic half-life (T1/2,int) and apparent intrinsic clearance (CLint,app) was also measured after incubation with human liver microsomes at 37.5 °C. In conclusion, BTZD derivatives may represent novel antidiabetics agents with fewer side effects than PPARγ full agonists.
Results
Chemistry
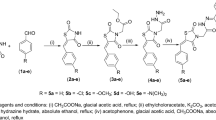
BTZDs (1–10) were prepared by Knoevenagel condensation of thiazolidinedione with appropriate benzaldehydes in the presence of piperidine as catalyst as previously reported40,41. 2-Chloroacetamide derivatives (a-k) were prepared by the reaction of 2-chloroacetylchloride with appropriate amines in the presence of triethylamine in chloroform42,43 (Fig. 1). Coupling of BTZDs with 2-chloroacetamides in the presence of triethylamine in acetonitrile provided final compounds. Known intermediates were confirmed by comparing their melting point with the literature value and final compounds were characterized by 1H-NMR, 13C-NMR and ESI-MS data.

Synthetic route for novel BTZD derviatives.
The structure of compound 1i was confirmed by X-ray crystallography (Fig. 2 and Table S1 of Supplementary Material). The compound crystallizes in the monoclinic P21/c space group with four molecules in the unit cell (a = 7.1761(4) Å, b = 26.1554(11) Å, c = 9.0667(4) Å, α = 90°, β = 112.127(3)°, γ = 90° and Z = 4). In the crystal structure, the TZD ring adopts a planar conformation with a maximum deviation of 0.0182(3) Å for C9. The mean planes of the thiazolidine and phenyl rings are inclined to one another by 5.74(7)°. The TZD and phenyl rings lie in plane, as evidenced from the torsion angles C6-C1-C7-C8 = 175.6° and C1-C7-C8-C9 = −178.5°. The oxygen atom (O1) is oriented syn-periplanar to C12 [C12-N1-C11-O1 = 5.1°] and anti-periplanar to C9 [C9-N1-C11-O1 = 179.7°]. The oxygen atom (O2) is oriented syn-periplanar to C7 [C7-C8-C9-O2 = −2.0°] and anti-periplanar to S1 [S1-C8-C9-O2 = 178.7°]. The carbonyl group (C13/O3) is oriented syn-periplanar to C14 [C14-N2-C13-O3 = −0.2°] and anti-periplanar to C16 [C16-N2-C13-O3 = −174.5°]. The 2-aminoacetamide moiety is in extended conformation, which can be seen from the torsion angle value of N1-C12-C13-N2 = −175.3°. Finally, the two torsion angles of the diethylamine group are C13-N2-C16-C17 = −104.9° and C13-N2-C14-C15 = −84.0°, respectively (Table S2 of Supplementary Material).

Molecular structure of 1i showing the atom labelling and displacement ellipsoids drawn at 30% probability level.
As shown in Fig. 2, molecule 1i has C-H···S and C-H···O intramolecular H-bonds forming a S(6) and S(5) ring motif. In addition, van der Waals forces also control the molecular packing, as shown in Fig. S2 of Supplementary Material. In the crystal, the molecules are stacked in layers, held together by π-π interactions, with Cg1 and Cg2 distance of 3.7435(13) Å (symmetry code Cg1 = x, y, z; Cg2 = 1 − x, −y, 2 − z.) between the centroids of two adjacent thiazolidine rings, as shown in Fig. S3 of Supplementary Material.
Transactivation induced by compounds 1a, 1i, and 3a as selective PPARγ partial agonists
A first batch of eleven compounds (1a-1k, Table 1) bearing an unsubstituted benzylidene moiety and different substituents on the acetamide nitrogen of TZD was evaluated for agonist activity on the human PPARα (hPPARα), PPARγ (hPPARγ), and PPARβ/δ (hPPARβ/δ) subtypes. For this purpose, GAL-4 PPAR chimeric receptors were expressed in transiently transfected HepG2 cells according to a previously reported procedure44,45. At first, the activity of these compounds was evaluated at two concentrations (5 μM and 25 μM) and compared with that of the corresponding reference agonists (Wy-14,643 for PPARα, Rosiglitazone for PPARγ and L-165,041 for PPARβ/δ) whose maximum induction was defined as 100%. Only 1a and 1i showed a weak to moderate selective activity towards PPARγ and, therefore, for these compounds a concentration-response curve was carried out. As displayed in Fig. 3A, their EC50 values were 7.0 ± 1.3 μM and 9.6 ± 0.8 μM with Emax 5.3 ± 0.3% and 23 ± 5%, respectively. In this figure, the concentration-response of Pioglitazone is also reported showing EC50 = 0.65 ± 0.01 μM and Emax = 80 ± 2%. Although weak or moderate, the PPARγ activity of 1a e 1i is not to be underestimated in that the well-known metaglidasen, having EC50 and Emax around 10 μM and 10% respectively, is still investigated as a useful agent for the treatment of T2DM and hyperglycemia, as demonstrated by some recent patents46,47. These results evidence the importance of flexible alkyl chains on the acetamide nitrogen atom of TZD moiety; in fact, all other derivatives present a rigid aliphatic or aromatic cycle that is probably unable to adapt its own conformation for accommodating into the PPARγ binding site. At this stage, with the aim to evaluate the influence of benzylidene moiety on PPAR activity, we introduced different substituents on the aromatic ring of 1a and 1i by preparing five analogues of the former (3a, 6a, 8a-10a) and nine analogues of the latter (2i-10i). These compounds (Table 2) were tested, again, at two concentrations (5 μM and 25 μM) towards all three PPAR subtypes. Only 3a, bearing a chlorine atom in the para position of benzylidene, exhibited a selective activity towards PPARγ and its concentration-response curve (Fig. 3A) provided EC50 = 5.9 ± 0.6 μM and Emax = 31.6 ± 1.0% values.

Characterization of 1a, 1i, and 3a binding to PPARγ. (A) Concentration-response curves of Rosiglitazone, Pioglitazone, 1a, 1i, and 3a in HepG2 cells co-transfected with pGal5TKpGL3 reporter and pGal4-hPPARγ-LBD. (B) SPR sensorgrams achieved injecting different concentrations (from 0.5 to 10 µM) of compounds 1a, 1i, and 3a on immobilized PPARγ. The positive control Rosiglitazone was injected at concentrations from 0.1 to 100 nM.
The ability of BTZDs 1a, 1i and 3a to interact with PPARγ was assessed by SPR-based Biacore binding assays, using Rosiglitazone as a positive control, as reported elsewhere48. Briefly, different concentrations (from 0.5 to 10 µM) of each compound were injected on a sensor chip, covalently modified by PPARγ. The resulting sensorgrams (Fig. 3B) showed that all the three compounds have some affinity towards PPARγ, as demonstrated by the measured KD of 38.3 ± 1.8 µM, 35.7 ± 2.6 µM and 32.0 ± 1.5 µM, respectively. However, these KD values are significantly lower than that measured for Rosiglitazone (320.5 ± 15.2 nM).
BTZDs 1a, 1i and 3a recognize PPARγ in a unique binding mode
To attempt to understand the molecular mechanism underlying the interaction of 1a, 1i and 3a with PPARγ and give an explanation to the significant increase of efficacy for 3a compared to 1a, we undertook docking studies into the PPARγ ligand-binding domain (LBD) using the GOLD Suite docking package49 in combination with the ChemPLP50 scoring function (rescoring with ChemScore)51. We selected the GQ-16/PPARγ crystal structure (PDB ID: 3T03)32 as a template for docking experiments, mainly based on the similarity of the structure features between our candidate compounds and GQ-16, both containing an BTZD portion. Analysis of the GQ-16/PPARγ complex revealed that the ligand is H-bonded via four water molecules to the protein: via W200 to Y327 (H5) and via W210, W211, and W61 to S342 (β-sheet), L340 (β-sheet), and R288 (H3). Accordingly, the four intervening water molecules were included in the docking experiments. Alternative conformations of F282 (H3), R288 (H3), F363 (H7) and Y473 (H12) side chains have been previously observed in PPARγ crystal structures complexed with several ligands, highlighting the innate plasticity of the PPARγ LBD52,53,54. Accordingly, the R288 side chain was allowed to move during the docking experiments. Validation test runs demonstrated that GOLD could reproduce the crystal bound conformation of GQ-16 and provide high alignment accuracy with low RMSD (0.74 Å, CHEMscore fitness = 33.8, CHEMscore ΔGbind = −39.8 kJ/mol).
Compounds 1a, 1i and 3a were found to fit well within the PPARγ binding pocket. However, they assumed a binding mode different from that of Rosiglitazone and other known PPARγ agonists. The binding mode of 3a as a representative example of all three compounds bound to PPARγ is shown in Fig. 4A. Although Rosiglitazone bind in a “U-shaped” conformation, wrapping around helix 3, our compounds bind in the LBD parallel to H3 (Fig. 4B). Given this binding mode, 1a, 1i and 3a do not contact the three key residues, H323 at helix 5, H449 at helix 11, and Y473 from the AF-2 helix in arm I of the LBD, whose interaction with the acidic head groups of TZDs may contribute to the receptor activation. Instead, they lie between H3 and the β-sheet (arms II and III of LBD), an area which is preferentially occupied by synthetic partial agonists such as PA-08254, MRL-24, nTZDpa, and BVT.1355. Three water molecules participate in an indirect H-bond network that involves both the TZD 4-oxo group and the amide C=O group of the N3-alkyl substituent of three ligands and the NH backbone of (β-sheet), the C=O backbone of L340 (β-sheet) and the NE atom of R288. The TZD 2-oxo group makes an indirect water-mediated interaction with the OH group of Y327, with a distance of 3.0 Å. The remaining interactions between ligands and protein are basically hydrophobic. In particular, the thiazolidinone ring contacts residues C285 (H3), L330 (H5), V339 (β-sheet), I341 (β-sheet), and M364 (H8). Significant interactions between the TZD sulfur atom with the sulfurs of the two specific residues C285 and M364 are at the distances 2.8 and 3.3 Å, respectively. The N3-alkyl substituents occupy the arm III of the PPARγ LBD cavity and make hydrophobic contacts with E295 (H3), A292 (H3), R288, M329 (H5), I326 (H5), L330 and L333 (H5). The benzylidene ring reaches into the pocket below H5 and forms hydrophobic interactions with I281 (H3), F282, C285, M348 (β-sheet), L353 (H6), L356 (H6/H7 loop), F360 (H7), F363 and M364 residues. Moreover, it engages in a parallel-displaced π-stacking interaction with the aromatic ring of F363 of H7. It is interesting to note that residues L356 and F360 appear in a suitable position to make lipophilic contacts with the p-chlorine atom of compound 3a, thus rationalizing the increase of efficacy of 3a compared to 1a and 1i.

In silico docking of compound 3a into the PPARγ binding pocket. (A) Binding mode of compound 3a (partial agonist, cyan sticks) into the PPARγ binding site represented as a dirtyviolet ribbon model. Only amino acids located within 4 Å of the bound ligand are displayed (white sticks) and labeled. H12 is shown in slate. H-bonds discussed in the text are depicted as dashed black lines. The water molecules bridging the interaction of the ligand with the protein are displayed as red spheres. (B) Cα superposition of the complexes of PPARγ with compound 3a and Rosiglitazone (a full agonist, magenta sticks, PDB code: 2PRG). The LBD of PPARγ consists of a hydrophobic entrance (arm III) that branches off into two subsites: the polar arm I, which is extended toward H12, and the hydrophobic arm II, which is located between helix H3 and β-sheet. The partial agonists occupy mainly arm II and arm III of the PPARγ LBD, but the full agonists occupy mainly arm I and arm II. (C) 2D ligand-interaction diagram of 3a. Positively charged amino acids are represented with dark blue circles, polar amino acids are represented with light blue circles and hydrophobic amino acids are represented with green circles. Hydrogen bonds are depicted with purple arrows–dashed arrows for H-bonds involving amino acid side chain and regular arrows for H-bonds involving amino acid backbone.
Cytotoxicity of 1a, 1i, and 3a against NIH/3T3 cells cells
The cytotoxic effects of BTZDs 1a, 1i and 3a on NIH-3T3 cells were subjected to cytotoxicity evaluation by XTT method as reported earlier56 (Fig. 5A). All three compounds were found having cytotoxicity at doses near or above 1000 µM (Table 3), but did not show any apparent toxicity at their effective doses. 3a was found to be the most potent among them, even though the three compounds shared very similar therapeutic values. According to cell viability data, 1a, 1i and 3a provided significantly higher viability than Rosiglitazone in vitro, especially at 316 and 1000 μM (Fig. 5A). At these concentrations, all of the compounds decreased cell viability in a dose-dependent manner. Also, 1i was the least cytotoxic compound among all of them. Therefore, 1a, 1i and 3a are valuable in terms of their safety and efficacy according to the results of in vitro studies.

Cell viability % of 1a, 1i and 3a against NIH/3T3 (A) and HepG2 (B) Cells. Data were analysed by two-way ANOVA followed by Tukey test and expressed as mean ± SEM. aindicates p < 0.05 vs 1000 μM; bindicates p < 0.05 vs 316 μM, cindicates p < 0.05 vs control; dindicates p < 0.05 vs 100 μM; eindicates p < 0.05 vs 10 μM; *indicates p < 0.05 vs Rosiglitazone; +indicates p < 0.05 vs 3a; #indicates p < 0.05 vs Pioglitazone.
Hepatotoxicity assessment of compounds 1a, 1i and 3a against HepG2 cells
Given that the first generation TZDs for T2DM treatment, like ciglitazone and troglitazone, were found to be highly hepatotoxic and thus showed limited clinical application and in consequence were rapidly withdrawn from the market after reports of severe liver failure and death57, a very critical point for PPARγ ligands to be considered as gold standard for T2DM drug discovery is that they did not present hepatotoxicity. For this reason, we investigated the hepatotoxicity of 1a, 1i and 3a on HepG2 cells in a concentration range of 0–1000 μM (Fig. 5B). HepG2 cells are considered as a useful model to study in vitro xenobiotic metabolism and liver toxicity. In particular, HepG2 cells are able to activate and detoxify xenobiotics and, they are the reason why these cells are intensively used in toxicology58,59. Hepatotoxicity was determined using the XTT assay. According to our results, all of the compounds had IC50 values greater than 1000 μM, which is far above their EC50 values. Among the tested compounds, Rosiglitazone and Pioglitazone were found to possess increased cell viability (Fig. 5B). At 1000 μM, 1a and 3a did not show any significant difference compared with Rosiglitazone and also reached higher viability values than Pioglitazone. Compound 1i only slightly reduced cell viability (63.97 ± 3.93%) at the highest tested concentration. Thus, 1a, 1i and 3a did not possess any hepatotoxicity risk at the tested concentrations, exhibiting a wide therapeutical safety range.
Antidiabetic activity of compounds 1a, 1i and 3a on STZ-NA-induced type 2 diabetic rats
PPARγ partial agonists 1a, 1i and 3a were investigated for their antidiabetic activity in a Streptozotocin-Nicotinamide (STZ-NA)-induced diabetic rat model. The test dose was fixed based on the predicted LD50 value60. A dose that is less than 1/20th of predicted LD50 was used for the study. Diabetic rats were orally treated with the test compounds at a dose of 36 mg/kg body weight for a period of 15 days. Fasting blood glucose of control, diabetic-untreated and diabetic-treated rats were tested on Days 1, 3, 7 and 15 of the treatment period (Fig. 6A).

In vivo antidiabetic activity of 1a, 1i, 3a in STZ-NA induced diabetic rats. Effects of compounds 1a, 1i, 3a, and Pioglitazone on the blood glucose levels (A), Hb (B), HbA1c (C), serum urea, creatinine, and uric acid (D), and TC, TG, LDL and VLDL (E). Data were analysed by one way ANOVA followed by Dunnett’s ‘t’ test and expressed as mean ± SEM from six observations; ***indicates p < 0.001 vs diabetic control, **indicates p < 0.01 vs diabetic control.
After administration for 15 days, the blood glucose levels of diabetic rats treated with compounds 1a, 1i, 3a, and Pioglitazone were significantly reduced compared with the blood glucose levels of the diabetic control rats. The results of in vitro study were consistent with in vivo results indicating that these compounds exerted the blood glucose lowering effect by activating PPARγ receptors. In particular, the data for 15th Day showed that the activity of compound 3a was equivalent to the standard drug Pioglitazone used in the study.
Total hemoglobin (Hb), Glycosylated hemoglobin (HbA1c), lipid profile [total cholesterol (TC), triglyceride (TG), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C) and very low-density lipoprotein cholesterol (VLDL-C)], urea, creatinine and uric acid levels were also measured before sacrificing the animals. Hb levels were significantly decreased in disease control (STZ + NA) when compared to normal control. 1a-, 1i-, 3a- and Pioglitazone-treated groups showed a significant increase in Hb levels (Fig. 6B). HbA1c is a reliable index of glycemic control in diabetes61. Glycosylation of Hb is directly proportional to fasting blood glucose levels62. In this study, STZ-NA-induced diabetic rats showed high levels of HbA1c compared to normal control. Administration of 1a, 1i and 3a significantly prevented the increase in HbA1c and this could be due to decrease in glucose levels (Fig. 6C). STZ-NA diabetic animals also displayed increased plasma levels of renal function parameters such as urea, creatinine, and uric acid, when compared to normal control. It was observed that all the active compounds significantly (p < 0.001) diminished the levels of urea, creatinine and uric acid compared to the mean values of diabetic control group (Fig. 6D). Diabetes mellitus is also associated with profound alterations in the plasma lipid profile63. An elevation in plasma concentrations of TG and LDL-C associated with marked reduction of VLDL-C are most typical feature of T2DM. Compared to normal control, disease control (STZ + NA) showed a significant increase in TC, TG, VLDL-C and LDL-C levels and a decrease in HDL-C level. The 1a-, 1i-, and 3a-treated groups showed an increase in HDL-C and a significant decrease in TC, T, VLDL-C, and LDL-C when compared to disease control (STZ + NA) (Fig. 6E). Collectively, these data clearly indicate the antidiabetic potential of BTZD derivatives 1a, 1i and 3a.
A histological study was carried out by using light microscopy in order to detect whether the treatment with 1a, 1i and 3a had an effect on the STZ-NA-induced diabetic pancreas and liver tissues. The histopathological examination of untreated diabetic rats showed a significant damage to pancreatic islets and degranulation of β-cells when compared to normal control pancreatic histology (Fig. 7), which had normal proportion of islets of Langerhans and acinar cells. The histological changes in diabetic rats were restored to near normal by treatments with 1a, 1i, 3a and Pioglitazone. In case of liver histopathological studies in experimental rats, the liver’s histological structure (Fig. 8A) was normal in the control group, with a central vein and surrounding hepatocytes. Figure 8B shows a liver with clear hepato-cellular necrosis and marked vacuolization with reducing nuclei size and disordered liver structure in STZ-NA-induced diabetic rats. Rats treated with 1a, 3a (Fig. 8C and E) and Pioglitazone (Fig. 8F) showed no hepatic abnormalities, and the arrangements of the hepatocytes in the liver were almost normal. In case of 1i-treated rats (Fig. 8E), the damage to the normal hepatocytes was observed more when compared with normal group.

Histopathological changes in the pancreatic islets of normal and STZ-NA-induced diabetic rats. (A) Pancreatic islet architecture of the normal control rats. (B) STZ-NA-induced diabetic control rats show pancreatic islet damage. (C) STZ-NA-induced diabetic rats treated with compound 1a (36 mg/kg body weight); (D) STZ-NA induced diabetic rats treated with compound 1i (36 mg/kg body weight); (E) STZ-NA-induced diabetic rats treated with compound 3a (36 mg/kg body weight); (F) STZ-induced diabetic rats treated with Pioglitazone (36 mg/kg body weight). Examinations were carried out at 40x magnifications with hematoxylin-eosin’s stain.

Histopathological changes in the liver of normal and STZ-NA-induced diabetic rats. (A) Normal Control. (B) Diabetic control. (C) STZ-NA-induced diabetic rats treated with compound 1a (36 mg/kg body weight); (D) STZ-NA-induced diabetic rats treated with compound 1i (36 mg/kg body weight); (E) STZ-NA-induced diabetic rats treated with compound 3a (36 mg/kg body weight); (F) STZ-induced diabetic rats treated with Pioglitazone (36 mg/kg body weight). Examinations were carried out at 40x magnifications with hematoxylin-eosin’s stain.
Pharmacokinetic profile of 1a, 1i and 3a
Aiming to identify if 1a, 1i and 3a could be suitable candidates for in vivo experiments, we profiled them in pharmacokinetic assays in vitro. The oral bioavailability of compounds in terms of Caco-2 and MDCK-II permeability and the P-glycoprotein (P-gp) efflux ratio were assessed64. The in vitro intrinsic half-life (T1/2,int) and the apparent intrinsic clearance (CLint,app) of compounds were also measured after incubation with human liver microsomes at 37.5 °C65. Apparent permeability coefficients (Papp) from A-to-B (apical to basolater of the cell monolayers) and B-to-A (basolateral to apical of the cell monolayers) were obtained by measuring the amount of the compound transported from the donor compartment at 2 μM to the receiver compartment after 150 min incubation. Quantification was done by LC-MS/MS. P-gp substrate activity was assessed by the efflux ratio (the ratio of Papp in the B-to-A direction over that in the A-to-B direction). As showed in Tables 4 and 5, compound 1i exhibited asymmetric transport across both Caco-2 and MDCK-II monolayers (Papp,A–B of 23.0 × 10−6 cm/sec for Caco-2 and 16.3 × 10−6 cm/sec for MDCK-II; Papp,B–A of 51.2 × 10−6 cm/sec for Caco-2 and 11.9 × 10−6 cm/sec for MDCK-II), suggesting that it may be orally absorbed in both mouse model and human studies. An efflux ratio (Papp,B–A/Papp,A–B) of >2 indicates that a compound may be a potential substrate for permeability glycoprotein (P-gp) or other active transporters. P-gp is an ATP-dependent transporter on the apical plasma membrane of enterocytes that functions to limit the entry of drugs into the cell. Compounds 1a, 1i and 3a showed efflux ratios less than 2.0 in MDCK-II cells, indicating that they are not potential substrates for P-gp and therefore have improved potential for oral absorption. In contrast, the efflux ratios were slightly higher than 2.0 in Caco-2 cells, suggesting the three compounds retained weak P-gp substrate activity.
Since the liver is the primary site of drug metabolism, subcellular fractions of liver microsomes are useful in vitro models of hepatic clearance as they still contain many of the drug metabolizing enzymes found in the liver (notably, the cytochrome P450 enzymes). Highly cleared compounds in vitro are likely to be rapidly cleared in vivo resulting in a short duration of action. In the human liver microsomal stability study, compound 1i was found to possess T1/2,int of 92.55 min, with an in vitro CLint,app of 18.71 µL/min/mg, which is an indicator of good metabolic stability (Table 6). In contrast, compound 1a exhibited low metabolic stability with T1/2,int of 10.20 min and CLint,app of 169.81 µL/min/mg.
Discussion
PPARγ is the pharmacological target of the insulin-sensitizing TZDs that have been widely used in the treatment of T2D. TZDs function as selective PPARγ ligands and induce transcription of PPARγ-targeted genes20. However, they are associated with various undesirable and severe side effects, including weight gain, fluid retention, edema, congestive heart failure, and bone fracture25,26,27. These limitations have raised substantial alarms and significantly compromised their future in many countries28. Therefore, it is critical to develop novel partial PPARγ agonists or SPPARγM that retain effective insulin-sensitizing action while minimizing the potential side effects, as demonstrated by recent studies in animal models and in clinical trials29,30,31,32,33,34.
In this study, we synthesized and gave the structural characteristics of a series of new BTZD derivatives bearing a substituent on the nitrogen atom of TZD nucleus, namely compounds 1a-1k, 2i-10i, 3a, 6a, and 8a-10a. Transactivation assays and SPR-based Biacore determinations indicated that compounds 1a, 1i, and 3a bound to PPARγ with low affinity compared with the well-characterized PPARγ agonists Rosiglitazone and Pioglitazone. The partial PPARγ agonist activity of these compounds may offer a distinct benefit because a number of studies have shown that partial PPARγ agonists, including selective SPPARγM, generate fewer side effects than full agonists29,30,31,32,33,34.
Docking experiments revealed that 1a, 1i, and 3a interact with PPARγ through a distinct binding mode, utilizing a north-south orientation, parallel to H3. Unlike full agonists, such as the TZDs or tyrosine analogs, BTZD derivatives show no direct H-bond interactions with the AF2 helix in arm I portion of the binding pocket but instead lie between H3 and the β-sheet, extending from arm II to arm III of the LBD. 1a, 1i, and 3a stabilize H3 and the β-sheet of PPARγ through water-mediated interactions between their TZD 4-oxo groups and both the NH backbone of S342 and the C=O backbone of L340. A further water-mediated H-bond is formed between the TZD 2-oxo group of ligands and the Y327 side chain. Moreover, compounds make numerous hydrophobic and van der Waals contacts with side chains of residues from H3, H5, H6 and H7.
Co-crystallography, mutagenesis and hydrogen/deuterium exchange (HDX) studies have all displayed that PPARγ full agonists activate the receptor through direct interactions with residues H323, H449 and Y47355,66,67,68. These interactions place the AF2 helix in a conformation that facilitates the recruitment of coactivator proteins and the activation of the transcriptional machinery. In contrast, partial agonists such as PA-08254, MRL-24, nTZDpa, and BVT.1355 activate PPARγ by a H12-independent mechanism55,69,70, i.e they do not bind to PPARγ by the network of H-bonds used by full agonists but preferentially stabilize other regions of the LBD, especially H3 and the β-sheet region, and/or promote modifications in the structure and dynamics of the flexible region known as Ω-loop71,72,73,74. This causes a lower degree of H12 stabilization, which affects the recruitment of coactivators and, in turn, decreases the transcriptional activity of PPARγ52,55. The lack of direct interaction between the H12 helix of the receptor and BTZD derivatives together with the stabilization of H3 and the β-sheet of PPARγ may explain the partial agonist/modulator activities of 1a, 1i, and 3a observed in vitro.
The results of our in vitro studies showed that compounds 1a, 1i, and 3a not only weakened toxicity on NIH-3T3 cells compared with the full PPARγ agonist Pioglitazone but also exhibited no hepatotoxicity risk at the tested concentrations. The antidiabetic and antilipidaemic activity of BTZDs were also evaluated by in vivo experiments in a STZ-NA-induced diabetic rat model. This disease model is characterized by moderate stable hyperglycemia, glucose intolerance, and reduced pancreatic insulin stores, and shares a number of syndromes with T2D patients75. In this study, administration of 1a, 1i, and 3a for 15 days in STZ-NA-induced diabetic rats significantly decreased serum blood glucose compared to control diabetic rats after 15 days of treatment. Compound 3a showed a glucose-lowering effect similar to Pioglitazone. Moreover, BTZDs-treated diabetic rats significantly decreased the level of HbA1C and increased Hb, which might be the result of an improvement in the glucose metabolism. Serum urea, uric acid and creatinine level demonstrate the role of 1a, 1i, and 3a in renal dysfunction. An augmented level of urea, uric acid and creatinine cause the osmotic diuresis and depletion of extracellular fluid volume in the diabetic condition in the serum of STZ-NA-induced diabetic rats76. 1a, 1i, and 3a treated groups showed a declined level of urea, uric acid, and creatinine, thus revealing a their positive effect in renal dysfunction.
It is well known that augmentation in TC and TG, LDL-C, VLDL-C, and the decline in the HDL-C are regarded as most common abnormalities in disease control (STZ + NA) group. In our study, the diabetic rats treated with 1a, 1i, and 3a showed an elevation in HDL-C and reduction in LDL-C and VLDL-C as evidenced by decreased levels of TC and TG, confirming the positive role of these compounds in increased insulin action on cholesterol and fatty acid biosynthesis and intestinal cholesterol absorption.
Histopathological findings of the pancreas and liver segments of BTZDs-treated diabetic rats revealed numerous pathological signs. Our findings demonstrated that the treatment of STZ-NA-induced diabetic rats with 1a, 1i and 3a restored to nearly normal the islets of Langerhans containing β-cells. Distorted central vein and hepatocytes were reinstated to normal in liver of STZ-NA-induced diabetic treated rats when compared to the toxic control. A good metabolic stability in liver microsomes, optimum Caco-2 and MDCK-II permeability, and pharmacokinetic profile in preclinical species are the major characteristics for a potential and safe drug molecule that could be further extrapolated in humans before clinical trials. In the present study, we tried to establish the in vitro pharmacokinetics, permeability, and efflux concerns for compounds 1a, 1i and 3a. Our in vitro findings indicated that 1i displays a good metabolic stability in human liver microsomes (T1/2,int = 92.55 min and CLint,app = 169.81 μL/min/mg), and a high Caco-2 and MDCK-II permeability with small efflux ratio. Taken together, as selective partial PPARγ agonists, BTZD derivatives 1a, 1i and 3a improved not only the homeostasis of glucose metabolism, but also that of the lipid metabolism, suggesting that these compounds are very promising pharmacological agents by selectively targeting PPARγ for further development in the clinical treatment of T2D.
Experimental Procedures
Synthesis of the BTZD derivatives
All the synthetic work was done procuring required laboratory and analytical grade chemicals, reagents and solvents. All reactions were carried out in oven-dried glassware under nitrogen atmosphere. Chemicals and solvents were of reagent grade and purchased from Sigma Aldrich/Merck/Spectro-chem/CDH. Completions of reactions were monitored on pre-coated TLC plates (silica gel 60 F-254, Merck™, KGaA, Germany). Melting points were determined on an OPTIMELT automated system apparatus and were uncorrected. Intermediates were characterized by their melting points. Compounds were purified by recrystallization using suitable solvents. Final compounds were characterized by their 1H NMR (300 MHz, ECX-500, JEOL and 400 MHz, VNMRS400), 13C NMR (400 MHz, VNMRS400), in either CDCl3 or DMSO-d 6 as a solvent. Mass spectra were recorded by WATERS-Q-T of Premier-HAB213 using the electrospray ionization-mass spectrometry (ESI-MS) technique. General procedure for the preparation of the BTZD derivatives, as well as NMR-H, NMR-C13 and ESI-MS spectra of compounds 1a-k, 3a, 6a, 8a-10a, 2i-10i were supplied in Supplementary Material (Figs. S4–S77).
Transcriptional transactivation assay
Reference compounds, the medium, and other cell culture reagents were purchased from Sigma-Aldrich (Milan, Italy).
Plasmids
The expression vectors expressing the chimeric receptor containing the yeast Gal4-DNA binding domain fused to either the human PPARα, PPARγ, or PPARδ LBD and the reporter plasmid for these Gal4 chimeric receptors (pGal5TKpGL3) containing five repeats of the Gal4 response elements upstream of a minimal thymidine kinase promoter that is adjacent to the luciferase gene were described previously77.
Cell culture and transfections
Human hepatoblastoma cell line HepG2 (Interlab Cell Line Collection, Genoa, Italy) was cultured in minimum essential medium (MEM) containing 10% of heat-inactivated foetal bovine serum, 100 U of penicillin G/mL, and 100 μg of streptomycin sulfate/mL at 37 °C in a humidified atmosphere of 5% CO2. For transactivation assays, 105 cells per well were seeded in a 24-well plate and transfections were performed after 24 h with CAPHOS, a calcium phosphate method, according to the manufacturer’s guidelines. Cells were transfected with expression plasmids encoding the fusion protein Gal4-PPARα-LBD, Gal4-PPARγ-LBD, or Gal4-PPARδ-LBD (30 ng), pGal5TKpGL3 (100 ng), and pCMVβgal (250 ng). Four hours after transfection, cells were treated for 20 h with the indicated ligands and reference compounds in duplicate. As a preliminary assay, all compounds were tested at two concentrations (5 and 25 μM); afterwards, only for ligands showing weak or moderate efficacy a concentration-response curve was carried out. Luciferase activity in cell extracts was determined by a luminometer (VICTOR3 V Multilabel Plate Reader, PerkinElmer). β-Galactosidase activity was determined using ortho-nitro-phenyl-β-D-galactopyranoside as described previously78. All transfection experiments were repeated at least twice.
Surface Plasmon Resonance
SPR analyses were carried out on a BIACORE 3000 instrument (GE-Healthcare). A PPARγ, surface was generated using a research-grade CM5 sensor chips (GE Healthcare). The protein (100 µg/ml in 10 mM CH3COONa, pH 5.0) was immobilized using a standard amine-coupling protocol, to obtain densities of 4 kRU. Testing compounds were dissolved in 100% DMSO to obtain 10 mM solutions, and then diluted in PBS containing variable amounts of DMSO, achieving a final DMSO concentration of 0.1%. For each molecule a five-point concentration series (0.5 µM, 1 µM, 2,5 µM, 5 µM and 10 µM) was set up. SPR experiments and data elaboration were carried out as reported elsewhere48.
Computational chemistry
Molecular modeling and graphics manipulations were performed using Maestro 10.5 (Schrödinger, LLC, New York, NY, 2016) and UCSF-CHIMERA 1.8.1 software packages, (http://www.cgl.ucsf.edu/chimera), running on an E4 Computer Engineering E1080 workstation E4 Computer Engineering E1080 workstation provided with an Intel Xeon processor. GOLD Suite 5.4.1 docking package (CCDC Software Limited: Cambridge, U.K., 2008)49 was used for all docking calculations. Figures were generated using Pymol 1.8.2 (Schrödinger, LLC, New York, NY, 2016).
Protein and Ligand Preparation
The coordinates of PPARγ in complex with GQ-16 (PDB code: 3T03)32, recovered from Brookhaven Protein Database79, were employed for the automated docking experiments. The protein setup was carried out using the Protein Preparation Wizard implemented in Maestro. Hydrogen atoms were added to the protein consistent with the neutral physiologic pH. Arginine and lysine side chains were considered as cationic at the guanidine and ammonium groups, and the aspartic and glutamic residues were considered as anionic at the carboxylate groups. The protonation and flip states of the imidazole rings of the histidine residues were adjusted together with the side chain amides of glutamine and asparagine residues in a H-bonding network optimization process. Successively, the protein hydrogens only were minimized using the Impref module of Impact with the OPLS_2005 force field. Initial coordinates of compounds 1a, 1i and 3a were constructed by using the Molecular Builder module in Maestro. The structures were energy-minimized using Macromodel 10.8 (Schrödinger, LLC, New York, NY, 2016) using the MMFF force field with the steepest descent (1000 steps) followed by truncated Newton conjugate gradient (500 steps) methods. Partial atomic charges were computed using the OPLS-AA force field.
Docking Simulations
Docking of 1a, 1i and 3a to PPARγ was performed with the GOLD software, which uses a genetic algorithm (GA) for determining the docking modes of ligands and proteins. The coordinates of the cocrystallized ligand GQ-16 were chosen as active-site origin. The active-site radius was set equal to 10 Å. Four explicit water molecules were allowed to toggle on or off during the individual docking runs (i.e., these waters were not automatically present in the binding site, but were included if their presence strengthened the interaction of the ligand with the receptor, as determined by the scoring function)80. Mobility of R288 side chain was established using the flexible side chains option in the GOLD front end, which incorporates the Lovell rotamer library81. Each GA run used the default parameters of 100 000 genetic operations on an initial population of 100 members divided into five subpopulations, with weights for crossover, mutation, and migration being set to 95, 95, and 10, respectively.
GOLD allows a user-definable number of GA runs per ligand, each of which starts from a different orientation. For these experiments, the number of GA runs was set to 1000 without the option of early termination, and scoring of the docked poses was performed with the original ChemPLP scoring function rescoring with ChemScore51. The final receptor-ligand complex for each ligand was chosen interactively by selecting the highest scoring pose that was consistent with the experimentally derived information about the binding mode of the ligand.
Cytotoxicity assessment
The 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) assay is based on the mitochondrial succinate dehydrogenase activity, which is only active in cells with an intact metabolism. XTT is reduced to soluble formazan by this enzyme and formazan is quantified and directly proportional to the number of viable cells. NIH/3T3 mouse embryonic fibroblast (ATCC®CRL-1658™) cell line was used to investigate the cytotoxicity of the compounds. NIH/3T3 cells were incubated according to the instructions of the supplier at 37 °C in a humidified atmosphere of 95% air and 5% CO2. NIH/3T3 cells were seeded at 1 × 104 cells into each well of 96-well plates. After 24 hours of incubation, compounds were added to the wells at the concentration range between 1 mM and 0,000316 mM concentrations (1000; 316; 100; 31.6; 10; 3.16; 1; 0.0316 µM) in quadruplicates. The XTT assay (Xenometrix, Allschwil, Switzerland) was performed according to the manufacturer’s instructions, after 24 h incubation with the compounds. The absorbance was determined after 2 h incubation at 480 nm with a reference wavelength of 680 nm using a microplate reader (BioTek, USA). Inhibition % was calculated for each concentration of the compounds according to the formula below obtained from the instructions of the manufacturer and IC50 values were estimated by plotting a dose response curve of the inhibition % versus test compound concentrations. IC50/EC50 values were calculated, values >1000 μM were accepted as 1000. Cell viability % was calculated according to growth control absorbance values.
The stock solutions of the compounds were prepared in DMSO and further dilutions were made with fresh culture medium. The final DMSO concentration was under 0.1%. Rosiglitazone and Pioglitazone were used as positive controls. All data were obtained from 3 independent experiments in quadruplicates.
In vitro hepatotoxicity assessment
In order to test the compounds for their hepatotoxicity, XTT assay was carried out on HepG2 human hepatocellular liver carcinoma cell line (ATCC® HB-8065™) as shown above. Cells were incubated according to the instructions of the supplier at 37 °C in a humidified atmosphere of 95% air and 5% CO2 and they were seeded at 1 × 104 cells into each well of 96-well plates. After 24 hours of incubation, eight different concentrations of the compounds were added to the wells (1000; 316; 100; 31.6; 10; 3.16; 1; 0.316 µM) in quadruplicates. The final DMSO concentration was set at <0.1%. Rosiglitazone and Pioglitazone were used as positive controls. IC50 values and cell viability % were calculated.
Animals and drug treatments
Male Wistar strain rats weighing about 160–200 g were used for animal studies. The experimental protocol was approved by the institutional animal ethical committee (no. PROV/BIT/PH/IAEC/04/2016) of the Department of Pharmaceutical Sciences and Technology, Birla Institute of Technology, Mesra, Ranchi, in accordance with National Institutes of Health Guide for Care and Use of Laboratory animals. Animals were housed in polypropylene cages containing wood shaving as bedding material, and maintained in the departmental animal house at 26 ± 2 °C and 44–55% relative humidity with a natural light/dark cycle, respectively. Rats were provided with rodent diet and water ad libitum. After initial duration of 12 h fast, the animals were rendered diabetic by a single intraperitonial administration of NA in normal saline at a dose of 230 mg/kg body weight, that was followed by freshly prepared solution of STZ in 0.1 M citrate buffer (pH 4.5) at a dose of 60 mg/kg body. The animals were allowed to drink 5% glucose solution overnight to prevent STZ-NA-induced hypoglycemia82,83. The rats were considered as diabetic if their blood glucose levels were above 250 mg/dL on the 3rd day after STZ-NA injection. The rats were divided into four groups of six animals in each group. Control animals received normal saline (Group I). Diabetic rats received STZ-NA injection (Group II). Diabetic rats orally fed with standard (Pioglitazone) and test drugs (1a, 1i and 3a) as 0.25% suspension in carboxymethyl cellulose at a dose of 36 mg/kg for 15 days (Groups III and IV). The blood glucose level of each group was checked at 1, 3, 7 and 15 Day by withdrawing blood from tail vein of the animal using glucose meter (ACCu-Chek active, Roche, Diagnostics USA).
Biochemical determinations
After pharmacological screening for antidiabetic activity, the animals were subjected to overnight fasting and blood was collected from retro-orbital region under light anaesthesia for performing various biochemical experiments. All the biochemical experiments were performed using commercially available kits. Total Hb was estimated by cyanomethaemoglobin method84, while HbA1c was estimated from whole blood using commercially available kits following the manufacturer guidelines (ERBA Diagnostics Inc, Accurex Biomedical Pvt. Ltd., Coral clinical systems, India). TG concentration was determined by GPO-POD enzymatic-colorimetric method85. TC level was measured by the end point, CHOD-POD colorimetric methods86. HDL-C was assayed by the method reported earlier87. VLDL-C and LDL-C in plasma were calculated by the Friedewald formula: VLDL-C = TG/5; LDL-C = TC-(HDL-C + VLDL-C)88. Levels of serum urea, uric acid, and creatinine were measured using commercially available kits89.
Histopathological study
Rats were sacrificed under light anesthesia using diethylether at the end of the pharmacological and biochemical studies. Pancreas and liver tissues were dissected, washed in ice cold physiological saline, fixed in a 15% buffered neutral formalin solution and, finally, embedded into paraffin blocks. Then, the tissue was sliced out into sections of 5 µm thickness by a rotator microtome and stained with hematoxylin-eosin. Obtained sections were examined by a Leica DME microscope, and representative photomicrographs were taken by a 7.1 megapixel Canon Power Shot S70 digital camera.
Pharmacokinetic profiling
The three active compounds 1a, 1i and 3a were subjected to in vitro Caco-2 and MDCK-II permeability studies, as well as metabolic stability assays based on earlier reported procedures64,65.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism 5.0 software for windows (San Diego, California, USA). Results were expressed as mean ± S.E.M. (standard error of the mean) for six rats in each group. The data was analysed by one way analysis of variance (ANOVA) followed by Dunnett’ multiple variance test. Differences were considered statistically significant at the levels of P < 0.05.
References
Shaw, J. E., Sicree, R. A. & Zimmet, P. Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes research and clinical practice 87, 4–14 (2010).
Hunt, K. J. & Schuller, K. L. The increasing prevalence of diabetes in pregnancy. Obstetrics and gynecology clinics of North America 34, 173–199 (2007).
Saltiel, A. R. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell 104, 517–529 (2001).
Viberti, G. Thiazolidinediones—Benefits on microvascular complications of type 2 diabetes. Journal of Diabetes and its Complications 19, 168–177 (2005).
Molitch, M. E. et al. Nephropathy in diabetes. Diabetes care 27, S79–83 (2004).
Hanefeld, M. et al. Risk factors for myocardial infarction and death in newly detected NIDDM: the Diabetes Intervention Study, 11-year follow-up. Diabetologia 39, 1577–1583 (1996).
Maccari, R. et al. Synthesis, induced-fit docking investigations, and in vitro aldose reductase inhibitory activity of non-carboxylic acid containing 2, 4-thiazolidinedione derivatives. Bioorganic & medicinal chemistry 16, 5840–5852 (2008).
Zhao, Z.-H. et al. Promoter methylation status and expression of PPAR-γ gene are associated with prognosis of acute-on-chronic hepatitis B liver failure. Clinical epigenetics 7, 1–9 (2015).
Kota, B. P., Huang, T. H.-W. & Roufogalis, B. D. An overview on biological mechanisms of PPARs. Pharmacological Research 51, 85–94 (2005).
Staels, B. et al. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 98, 2088–2093 (1998).
Elisaf, M. Effects of fibrates on serum metabolic parameters. Curr Med Res Opin 18, 269–276 (2002).
Sprecher, D. L. L. Lipoproteins, and peroxisome proliferator activated receptor-delta. Am J Cardiol 100, n20–24, https://doi.org/10.1016/j.amjcard.2007.08.009 (2007).
Reilly, S. M. & Lee, C. H. PPAR delta as a therapeutic target in metabolic disease. FEBS Lett 582, 26–31, https://doi.org/10.1016/j.febslet.2007.11.040 (2008).
Pirat, Cl. et al. Targeting peroxisome proliferator-activated receptors (PPARs): development of modulators. J. Med. Chem. 55, 4027–4061 (2012).
Fajas, L., Fruchart, J.-C. & Auwerx, J. PPARγ3 mRNA: a distinct PPARγ mRNA subtype transcribed from an independent promoter. FEBS letters 438, 55–60 (1998).
Tontonoz, P. & Spiegelman, B. M. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem 77, 289–312, https://doi.org/10.1146/annurev.biochem.77.061307.091829 (2008).
Lehrke, M. & Lazar, M. A. The many faces of PPARγ. Cell 123, 993–999 (2005).
Daynes, R. A. & Jones, D. C. Emerging roles of PPARs in inflammation and immunity. Nature Reviews Immunology 2, 748–759 (2002).
Berger, J. P., Akiyama, T. E. & Meinke, P. T. PPARs: therapeutic targets for metabolic disease. Trends in pharmacological sciences 26, 244–251 (2005).
Cheng, P. T. & Mukherjee, R. PPARs as targets for metabolic and cardiovascular diseases. Mini reviews in medicinal chemistry 5, 741–753 (2005).
Fujita, T. et al. Reduction of insulin resistance in obese and/or diabetic animals by 5-[4-(1-methylcyclohexylmethoxy) benzyl]-thiazolidine-2, 4-dione (ADD-3878, U-63,287, ciglitazone), a new antidiabetic agent. Diabetes 32, 804–810 (1983).
Parulkar, A. A., Pendergrass, M. L., Granda-Ayala, R., Lee, T. R. & Fonseca, V. A. Nonhypoglycemic effects of thiazolidinediones. Annals of Internal Medicine 134, 61–71 (2001).
Nissen, S. E. & Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine 356, 2457–2471 (2007).
Kim, J. W. et al. Tolerability and pharmacokinetics of lobeglitazone (CKD-501), a peroxisome proliferator-activated receptor-γ agonist: a single-and multiple-dose, double-blind, randomized control study in healthy male Korean subjects. Clinical therapeutics 33, 1819–1830 (2011).
Grey, A. Skeletal consequences of thiazolidinedione therapy. Osteoporos Int 19, 129–137, https://doi.org/10.1007/s00198-007-0477-y (2008).
Rubenstrunk, A., Hanf, R., Hum, D. W., Fruchart, J. C. & Staels, B. Safety issues and prospects for future generations of PPAR modulators. Biochim Biophys Acta 1771, 1065–1081, https://doi.org/10.1016/j.bbalip.2007.02.003 (2007).
Shearer, B. G. & Billin, A. N. The next generation of PPAR drugs: do we have the tools to find them? Biochim Biophys Acta 1771, 1082–1093, https://doi.org/10.1016/j.bbalip.2007.05.005 (2007).
Jones, D. Potential remains for PPAR-targeted drugs. Nat Rev Drug Discov 9, 668–669, https://doi.org/10.1038/nrd3271 (2010).
Berger, J. P. et al. Distinct properties and advantages of a novel peroxisome proliferator-activated protein [gamma] selective modulator. Mol Endocrinol 17, 662–676, https://doi.org/10.1210/me.2002-0217 (2003).
Rocchi, S. et al. A unique PPARgamma ligand with potent insulin-sensitizing yet weak adipogenic activity. Mol Cell 8, 737–747 (2001).
Choi, J. H. et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466, 451–456, https://doi.org/10.1038/nature09291 (2010).
Amato, A. A. et al. GQ-16, a novel peroxisome proliferator-activated receptor gamma (PPARgamma) ligand, promotes insulin sensitization without weight gain. J Biol Chem 287, 28169–28179, https://doi.org/10.1074/jbc.M111.332106 (2012).
Choi, J. H. et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature 477, 477–481, https://doi.org/10.1038/nature10383 (2011).
Lee, D. H., Huang, H., Choi, K., Mantzoros, C. & Kim, Y. B. Selective PPARgamma modulator INT131 normalizes insulin signaling defects and improves bone mass in diet-induced obese mice. Am J Physiol Endocrinol Metab 302, E552–560, https://doi.org/10.1152/ajpendo.00569.2011 (2012).
Zhou, L. et al. Design, synthesis and evaluation of PPAR gamma binding activity of 2-thioxo-4-thiazolidinone derivatives. Chinese Chemical Letters 26, 63–68 (2015).
Lu, J. et al. Design, Synthesis, and Activity Evaluation of GK/PPARγ Dual‐Target‐Directed Ligands as Hypoglycemic Agents. ChemMedChem 9, 922–927 (2014).
Colca, G. R., Kletzien, R. F., Tanis, S. P. & Larsen, S. D. Inventors; Metabolic Solutions Development Company, assignee. PPAR-sparing thiazolidinediones and combinations for the treatment of neurodegenerative diseases. United States patent WO2011084456 A1. 2011 July 14.
Sha, X., Zhang, H., Huang, W., Zhou, J. & Zhou, Y. Design and Synthesis of Thiazolidinedione Derivates as Selective PPARγ Modulators. Letters in Drug Design & Discovery 6, 224–227 (2009).
Madhavan, G. R. et al. Dual PPAR-α and-γ activators derived from novel benzoxazinone containing thiazolidinediones having antidiabetic and hypolipidemic potential. Bioorganic & medicinal chemistry 14, 584–591 (2006).
Bruno, G. et al. Synthesis and aldose reductase inhibitory activity of 5-arylidene-2, 4-thiazolidinediones. Bioorganic & medicinal chemistry 10, 1077–1084 (2002).
Kar, K. et al. Design, synthesis and glucose uptake activity of some novel glitazones. Bioorganic chemistry 56, 27–33 (2014).
Jöst, C., Nitsche, C., Scholz, T., Roux, L. & Klein, C. D. Promiscuity and selectivity in covalent enzyme inhibition: a systematic study of electrophilic fragments. Journal of medicinal chemistry 57, 7590–7599 (2014).
Joshi, M. C. et al. In vitro antimalarial activity, β-haematin inhibition and structure–activity relationships in a series of quinoline triazoles. European journal of medicinal chemistry 69, 338–347 (2013).
Pinelli, A. et al. Synthesis, biological evaluation, and molecular modeling investigation of new chiral fibrates with PPARα and PPARγ agonist activity. Journal of medicinal chemistry 48, 5509–5519 (2005).
Porcelli, L. et al. Synthesis, characterization and biological evaluation of ureidofibrate-like derivatives endowed with peroxisome proliferator-activated receptor activity. Journal of medicinal chemistry 55, 37–54 (2011).
Rauch, T., Hamilton, B. S. & Tsutsumi, M. Inventors; Boehringer Ingelheim International Gmbh, assignee. Pharmaceutical combinations for the treatment of metabolic disorders. United States patent WO2013167554 A1. 2013 November 14.
Hamilton, B. S., Rauch, T. & Tsutsumi, M. Inventors; Boehringer Ingelheim International Gmbh, assignee. Pharmaceutical combinations for the treatment of metabolic disorders. United States patent WO2012059416 A1. 2012 May 10.
Vassallo, A., Vaccaro, M. C., De Tommasi, N., Dal Piaz, F. & Leone, A. Identification of the plant compound geraniin as a novel Hsp90 inhibitor. PLoS One 8, e74266, https://doi.org/10.1371/journal.pone.0074266 (2013).
Jones, G., Willett, P., Glen, R. C., Leach, A. R. & Taylor, R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267, 727–748, https://doi.org/10.1006/jmbi.1996.0897 (1997).
Verdonk, M. L. et al. Docking performance of fragments and druglike compounds. J Med Chem 54, 5422–5431, https://doi.org/10.1021/jm200558u (2011).
Verdonk, M. L., Cole, J. C., Hartshorn, M. J., Murray, C. W. & Taylor, R. D. Improved protein-ligand docking using GOLD. Proteins 52, 609–623, https://doi.org/10.1002/prot.10465 (2003).
Lu, I. L. et al. Structure-based drug design of a novel family of PPARgamma partial agonists: virtual screening, X-ray crystallography, and in vitro/in vivo biological activities. J Med Chem 49, 2703–2712, https://doi.org/10.1021/jm051129s (2006).
Ebdrup, S. et al. Synthesis and biological and structural characterization of the dual-acting peroxisome proliferator-activated receptor alpha/gamma agonist ragaglitazar. J Med Chem 46, 1306–1317, https://doi.org/10.1021/jm021027r (2003).
Burgermeister, E. et al. A novel partial agonist of peroxisome proliferator-activated receptor-gamma (PPARgamma) recruits PPARgamma-coactivator-1alpha, prevents triglyceride accumulation, and potentiates insulin signaling in vitro. Mol Endocrinol 20, 809–830, https://doi.org/10.1210/me.2005-0171 (2006).
Bruning, J. B. et al. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure 15, 1258–1271, https://doi.org/10.1016/j.str.2007.07.014 (2007).
Kwiecińska, P., Taubøll, E. & Gregoraszczuk, E. Ł. Effects of valproic acid and levetiracetam on viability and cell cycle regulatory genes expression in the OVCAR-3 cell line. Pharmacological Reports 64, 157–165 (2012).
Guo, L. et al. Differences in hepatotoxicity and gene expression profiles by anti-diabetic PPAR gamma agonists on rat primary hepatocytes and human HepG2 cells. Mol Divers 10, 349–360, https://doi.org/10.1007/s11030-006-9038-0 (2006).
Gomez-Lechon, M. J., Tolosa, L. & Donato, M. T. Upgrading HepG2 cells with adenoviral vectors that encode drug-metabolizing enzymes: application for drug hepatotoxicity testing. Expert Opin Drug Metab Toxicol 13, 137–148, https://doi.org/10.1080/17425255.2017.1238459 (2017).
Garcia-Canaveras, J. C. et al. LC-MS untargeted metabolomic analysis of drug-induced hepatotoxicity in HepG2 cells. Electrophoresis, https://doi.org/10.1002/elps.201500095 (2015).
Pires, D. E., Blundell, T. L. & Ascher, D. B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. Journal of medicinal chemistry 58, 4066–4072 (2015).
SubashBabu, P., Prabuseenivasan, P. & Ignacimuthu, S. (2007).
Babu, P. S., Prabuseenivasan, S. & Ignacimuthu, S. Cinnamaldehyde—a potential antidiabetic agent. Phytomedicine 14, 15–22 (2007).
Dunn, F. L. Hyperlipidemia in diabetes mellitus. Diabetes/metabolism reviews 6, 47–61 (1990).
Kellard, L. & Engelstein, M. Automation of cell-based and noncell-based permeability assays. JALA: Journal of the Association for Laboratory Automation 12, 104–109 (2007).
Mondal, S., Mazumder, U., Mondal, N. & Banerjee, S. Optimization of rat liver microsomal stability assay using HPLC. J Biol Sci 8, 1110–1114 (2008).
Hamuro, Y. et al. Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci 15, 1883–1892, https://doi.org/10.1110/ps.062103006 (2006).
Nolte, R. T. et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 395, 137–143, https://doi.org/10.1038/25931 (1998).
Chalmers, M. J., Busby, S. A., Pascal, B. D., Southern, M. R. & Griffin, P. R. A two-stage differential hydrogen deuterium exchange method for the rapid characterization of protein/ligand interactions. J Biomol Tech 18, 194–204 (2007).
Hughes, T. S. et al. Ligand and receptor dynamics contribute to the mechanism of graded PPARgamma agonism. Structure 20, 139–150, https://doi.org/10.1016/j.str.2011.10.018 (2012).
Kroker, A. J. & Bruning, J. B. Review of the Structural and Dynamic Mechanisms of PPARgamma Partial Agonism. PPAR Res 2015, 816856, https://doi.org/10.1155/2015/816856 (2015).
Waku, T. et al. Structural insight into PPARgamma activation through covalent modification with endogenous fatty acids. J Mol Biol 385, 188–199, https://doi.org/10.1016/j.jmb.2008.10.039 (2009).
Waku, T., Shiraki, T., Oyama, T. & Morikawa, K. Atomic structure of mutant PPARgamma LBD complexed with 15d-PGJ2: novel modulation mechanism of PPARgamma/RXRalpha function by covalently bound ligands. FEBS Lett 583, 320–324, https://doi.org/10.1016/j.febslet.2008.12.017 (2009).
Puhl, A. C. et al. Mode of peroxisome proliferator-activated receptor gamma activation by luteolin. Mol Pharmacol 81, 788–799, https://doi.org/10.1124/mol.111.076216 (2012).
Vasaturo, M. et al. A compound-based proteomic approach discloses 15-ketoatractyligenin methyl ester as a new PPARgamma partial agonist with anti-proliferative ability. Sci Rep 7, 41273, https://doi.org/10.1038/srep41273 (2017).
Masiello, P. et al. Experimental NIDDM: development of a new model in adult rats administered streptozotocin and nicotinamide. Diabetes 47, 224–229 (1998).
Sheela, N., Jose, M. A., Sathyamurthy, D. & Kumar, B. N. Effect of silymarin on streptozotocin-nicotinamide-induced type 2 diabetic nephropathy in rats. Iran J Kidney Dis 7, 117–123 (2013).
Raspe, E. et al. Modulation of rat liver apolipoprotein gene expression and serum lipid levels by tetradecylthioacetic acid (TTA) via PPARalpha activation. J Lipid Res 40, 2099–2110 (1999).
Hollon, T. & Yoshimura, F. K. Variation in enzymatic transient gene expression assays. Anal Biochem 182, 411–418 (1989).
Bernstein, F. C. et al. The Protein Data Bank. A computer-based archival file for macromolecular structures. Eur J Biochem 80, 319–324 (1977).
Verdonk, M. L. et al. Modeling water molecules in protein-ligand docking using GOLD. J Med Chem 48, 6504–6515, https://doi.org/10.1021/jm050543p (2005).
Lovell, S. C. et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50, 437–450, https://doi.org/10.1002/prot.10286 (2003).
Nazreen, S. et al. Thiazolidine-2,4-diones derivatives as PPAR-γ agonists: Synthesis, molecular docking, in vitro and in vivo antidiabetic activity with hepatotoxicity risk evaluation and effect on PPAR-γ gene expression. Bioorganic & medicinal chemistry letters 24, 3034–3042 (2014).
Altan, N. et al. Effect of the sulfonylurea glyburide on superoxide dismutase in streptozotocin-induced diabetic rat muscle. General Pharmacology: The Vascular System 28, 795–796 (1997).
Drabkin, D. L. & Austin, J. H. Spectrophotometric studies I. Spectrophotometric constants for common hemoglobin derivatives in human, dog, and rabbit blood. Journal of Biological Chemistry 98, 719–733 (1932).
McGowan, M., Artiss, J. D., Strandbergh, D. R. & Zak, B. A peroxidase-coupled method for the colorimetric determination of serum triglycerides. Clinical chemistry 29, 538–542 (1983).
Roeschlau, P., Bernt, E. & Gruber, W. Enzymatic determination of total cholesterol in serum. Zeitschrift fur klinische Chemie und klinische Biochemie 12, 226–226 (1974).
Grove, T. H. Effect of reagent pH on determination of high-density lipoprotein cholesterol by precipitation with sodium phosphotungstate-magnesium. Clinical Chemistry 25, 560–564 (1979).
Friedewald, W. T., Levy, R. I. & Fredrickson, D. S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clinical chemistry 18, 499–502 (1972).
Jaffé, M. Ueber den Niederschlag, welchen Pikrinsäure in normalem harn erzeugt und über eine neue reaction des kreatinins. Zeitschrift für physiologische Chemie 10, 391–400 (1886).
Acknowledgements
First author acknowledge UGC-MANF (MUS-JH-12-13-11472) for award of JRF fellowship. We are also thankful to DST-FIST (SR/FST/CSI-242/2012) for NMR facility at Birla Institute of Technology, Mesra and Institute of Life Sciences, Hyderabad, AP, India for providing spectral data.
Author information
Authors and Affiliations
Contributions
V.J. and A. Lavecchia designed the study. V.J. and S.Y. made the synthesis of compounds. F.L. and A. Laghezza made the biological test. F.D.P. made the SPR experiments. A. Lavecchia and F.C. performed the docking experiments. V.D. and V.V. made the crystallographic study. A.K.P. and S.Y. made the in vivo pharmacology experiments and the histopathological studies. O.A. and M.B. performed the in vitro cytotoxicity studies. S.K.M. performed the pharmacokinetic study. V.J. and A. Lavecchia wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Accession Codes: The crystallographic data of compound 1i described here have been deposited in the Cambridge Crystallographic Data Center (CCDC) database under accession number 1501668. This data can be obtained free of charge from the CCDC via www.ccdc.cam.ac.uk/data_request/cif.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yasmin, S., Capone, F., Laghezza, A. et al. Novel Benzylidene Thiazolidinedione Derivatives as Partial PPARγ Agonists and their Antidiabetic Effects on Type 2 Diabetes. Sci Rep 7, 14453 (2017). https://doi.org/10.1038/s41598-017-14776-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14776-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
