
Abstract
Sialylation regulates the in vivo half-life of recombinant therapeutic glycoproteins, affecting their therapeutic efficacy. Levels of the precursor molecule cytidine monophospho-N-acetylneuraminic acid (CMP-Neu5Ac) are considered a limiting factor in the sialylation of glycoproteins. Here, we show that by reducing the amount of intracellular CMP-Neu5Ac consumed for glycosphingolipid (GSL) biosynthesis, we can increase the sialylation of recombinant human erythropoietin (rhEPO) produced in CHO cells. Initially, we found that treating CHO cells with a potent inhibitor of GSL biosynthesis increases the sialylation of the rhEPO they produce. Then, we established a stable CHO cell line that produces rhEPO in the context of repression of the key GSL biosynthetic enzyme UDP-glucose ceramide glucosyltransferase (UGCG). These UGCG-depleted cells show reduced levels of gangliosides and significantly elevated levels of rhEPO sialylation. Upon further analysis of the resulting N-glycosylation pattern, we discovered that the enhanced rhEPO sialylation could be attributed to a decrease in neutral and mono-sialylated N-glycans and an increase in di-sialylated N-glycans. Our results suggest that the therapeutic efficacy of rhEPO produced in CHO cells can be improved by shunting intracellular CMP-Neu5Ac away from GSL biosynthesis and toward glycoprotein sialylation.
Similar content being viewed by others

Introduction
Many of the therapeutic bio-drugs currently in commercial production are sialylated glycoproteins1. Sialic acid is a terminal sugar residue on the N- and O-linked glycosylation chains attached to many glycoproteins. Since secreted therapeutic glycoproteins are degraded by hepatocytes after being recognized by the asialoglycoprotein receptor (ASGPR), enhanced sialylation of these therapeutic glycoproteins can increase their in vivo circulating half-life2,3. Thus, because it will likely enhance therapeutic efficacy, there is considerable interest in developing methods for enhancing the sialylation of recombinant glycoproteins.
Early attempts at promoting sialylation focused on the over-expression of sialylation-related enzymes4,5, but these efforts quickly hit a ceiling6. It became clear that the intracellular level of CMP-Neu5Ac, a precursor required for sialic acid biosynthesis, was limiting7,8,9. Attempts to increase intracellular CMP-Neu5Ac have included supplementation of the CMP-Neu5Ac biosynthetic precursor ManNAc10, over-expression of the CMP-Neu5Ac transporter (CST)11,12, and over-expression of mutant UDP-N-acetyl-glucosamine-2-epimerase (GNE) to bypass CMP-Neu5Ac-mediated feedback inhibition13,14. Recently, we also found a further increase in rhEPO sialylation upon over-expression of α2,3-sialyltransferase (ST) in CHO cells expressing mutant GNE and CST15.
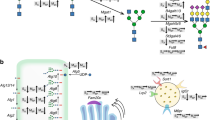
In addition to its role in glycoprotein biology, sialic acid is also attached to glycosphingolipids (GSLs) including those of the Ganglio-series (GlcNAcβ4Galβ4GlcβCer), Globo-series (Galα4Galβ4GlcβCer), Lacto-series (GlcNAcβ3Galβ4GlcβCer), and Neolacto-series (Galβ4GlcNAcβ3Galβ4GlcβCer)16. Sialylation occurs when CMP-Neu5Ac is translocated to the Golgi complex. There, ST catalyzes the transfer of Neu5Ac to a terminal galactosyl residue17. The inhibition of CMP-Neu5Ac translocation to the Golgi reduces the sialylation of both glycoproteins and GSLs, suggesting CMP-Neu5Ac is required for both18. It is unclear, however, whether GSL sialylation would correlate with glycoprotein sialylation. This is the question we attempted to address in the present study. Indeed, we found that treating CHO cells with a potent inhibitor of GSL biosynthesis increases their sialylation of rhEPO. Subsequently, we established a stable cell line with reduced expression of the UGCG gene. These cells too show enhanced rhEPO sialylation. After performing a more detailed N-glycan analysis, we found that UGCG-depleted CHO cells produce rhEPO with increased levels of di-sialylated N-glycan.
Results
EtDO-P4 treatment enhances sialylation
To investigate the effect of GSL biosynthesis on glycoprotein sialylation, we used a chemical inhibitor of GSL biosynthesis. Ceramide glucosyltransferase (CGT) catalyzes the transfer of the glucose moiety from UDP-glucose to ceramide to produce glucosylceramide (GlcCer). GlcCer, in turn, acts as the GSL core (Fig. 1). EtDO-P4 (D-threo-ethylenedioxyphenyl-2-palmitoylamino-3-pyrrolidino-propanol) inhibits CGT and is, therefore, capable of reducing GSL biosynthesis in various cells19,20. After treating rhEPO-producing EC2-1H9 CHO cells with EtDO-P4, we measured their GSL biosynthesis using thin-layer chromatography (TLC) as previously described21. We found that ganglioside GM3 is the predominant GSL in CHO cells, with gangliosides GM1 and GM2 making up only minor fractions (Fig. 2A). After EtDO-P4 treatment, however, we observed a significant reduction in GSL biosynthesis (Fig. 2A). Although ganglioside GM3 reportedly induces cell proliferation in various cancer cells by activating cell surface receptors like EGFR and integrins22, EtDO-P4 does not alter cell proliferation in EC2-1H9 CHO cells (Fig. S1).

Ganglioside biosynthesis schematic. UGCG catalyzes the first step of GSL biosynthesis. CMP-Neu5Ac is a sialylation precursor in both glycoproteins and GSLs.

EtDO-P4 treatment increases rhEPO sialylation. (A) Total GSLs extracted from cells were subjected to TLC followed by staining with orcinol/sulfuric acid. Lane 1, GSL standards; Lane 2, GSLs extracted from EC2-1H9 cells; Lane 3, GSLs extracted from EC2-1H9 treated with EtDO-P4. (B) rhEPO was purified using immunoprecipitation with an anti-EPO antibody. Sialylation was determined using WGA lectin. (C) Quantification of data in B; n = 3. **P < 0.01 (Two-tailed Student’s t-test). Data are presented as means ± S.E.M.
We previously reported that over-expression of glycosylation enzymes in CHO cells producing rhEPO leads to increased rhEPO sialylation6,23. To determine whether the inhibition of GSL biosynthesis can enhance the sialylation of therapeutic glycoproteins, we cultured EC2-1H9 CHO cells producing rhEPO as previously described23. We then purified rhEPO they produced via immunoprecipitation with an anti-EPO antibody and measured its sialylation level using lectin blotting with wheat germ agglutinin (WGA). This technique is specific for terminal sialic acids24. Indeed, we found that although EtDO-P4 treatment slightly reduces rhEPO production, it increases rhEPO sialylation by 50% (Fig. 2B and C). This suggests reducing the amount of CMP-Neu5Ac consumed by GSL production increases rhEPO sialylation.
Transient inhibition of GSL biosynthesis enhances rhEPO sialylation
From a commercial perspective, the treatment of cells with chemical inhibitors is generally considered impractical because of the cost and potential for side effects. Instead, we hoped to develop a stable cell line that produces highly sialylated rhEPO as a consequence of its reduced expression of UGCG. As a first step in this direction, we designed three different small interfering RNAs to specifically target UGCG (Table SI). We were able to confirm that all three siRNAs significantly repress the expression of UGCG by EC2-1H9 CHO cells (Fig. S2A). Consistent with what we observed with EtDO-P4 treatment, all three siRNAs reduce the levels of ganglioside GM3 (Fig. 3A) and other GSLs (Fig. 3B). We next used an anti-EPO antibody to pull-down rhEPO from supernatant collected from these UGCG-depleted EC2-1H9 CHO cells. As expected, we found rhEPO purified from these UGCG-depleted CHO cells shows 50% more sialylation than rhEPO obtained from control cells (Fig. 3C and D).

Transient repression of UGCG increases rhEPO sialylation. (A) Total GSLs extracted from cells were subjected to TLC followed by staining with orcinol/sulfuric acid. Lane 1, GM3 standard; Lane 2, GSLs extracted from EC2-1H9 cells; Lanes 3–5, GSLs extracted from EC2-1H9 cells treated with siRNAs #1–3; n = 3. (B) Quantification of total GSLs. n = 3. (C) rhEPO was purified using immunoprecipitation with an anti-EPO antibody. Sialylation was determined using MAL I lectin. (D) Quantification of data in (C); n = 3. *P < 0.05, **P < 0.01 (Two-tailed Student’s t-test). Data are presented as means ± S.E.M.
Stable UGCG depletion also enhances rhEPO sialylation
Since transient knock-down of UGCG enhances rhEPO sialylation, we next attempted to establish a cell line that produces rhEPO in the context of stable UGCG depletion. All three siRNAs produce similar levels of rhEPO sialylation (see Fig. 3), so we designed an artificial miRNA expression vector, pcUGCG-miR-1, to target the same sequence as siRNA1. We transfected this vector into EC2-1H9 CHO cells and selected for a stable cell line. We confirmed that expression of the artificial miRNA reduces expression of UGCG as compared to control EC2-1H9 CHO cells (Fig. S2B). We also found that enzymatic activity of UGCG was inhibited in UGCG depleted cells as examined by the conversion of NBD-ceramide to NBD-glucosylceramide (Fig. 4A). The stable CHO cell line, which we named EC2-1H9-miUGCG, shows significantly reduced levels of ganglioside GM3 as well as other GSLs (Fig. 4B and C). Importantly, we confirmed that the rate of proliferation of EC2-1H9-miUGCG cells is similar to that of EC2-1H9 cells (Fig. 4D).

microRNA-mediated repression of UGCG in CHO cells. (A) Enzymatic activity of UGCG was determined by conversion of NBD-Cer to NBD-GlcCer on TLC. Left, Lane 1, Standard lipids; Lane 2, lipids extracted from control cells; Lane 3, lipids extracted from UGCG depleted cells. Right, Quantification of lipids was shown. (B) Total GSLs extracted from cells were subjected to TLC followed by staining with orcinol/sulfuric acid. Lane 1, GM3 standard; Lane 2, GSLs extracted from EC2-1H9 cells; Lane 3, GSLs extracted from EC2-1H9-miUGCG cells. n = 3. (C) Quantification of total GSLs. n = 3, **P < 0.01 (Two-tailed Student’s t-test). (D) Cell proliferation was determined for 6 days of culture. Closed circles, EC2-1H9 cells; Open circles, EC2-1H9-miUGCG. n = 3. Data are presented as means ± S.E.M.
To determine whether stable UGCG depletion enhances rhEPO sialylation, we allowed the EC2-1H9-miUGCG cells to produce rhEPO for 4 days (see Methods). We then used an anti-EPO antibody to purify rhEPO from the resulting supernatant using immunoaffinity chromatography. Both the EC2-1H9-miUGCG cells and the control EC2-1H9 cells produce similar levels of rhEPO (Fig. 5A). Although both types of cells show similar levels of intracellular CMP-Neu5Ac, EC2-1H9-miUGCG cells produce rhEPO with significantly more sialylation than that produced by EC2-1H9 cells (Fig. 5B and C). The similar levels of intracellular CMP-Neu5Ac suggest that the concentration of this metabolite in the sialylation pathway is maintained in a state of dynamic equilibrium. Since rhEPO contains one O-glycosylation site (Ser 126) and three N-glycosylation sites (Asn 24, Asn 38, and Asn 83), a single mole of rhEPO can be decorated with up to 14 moles of sialic acid25. Previously, we measured rhEPO sialylation using an o-phenylenediamine (OPD)-labeling technique coupled with reversed-phase HPLC with a C18 column15,26. Using this same approach, we found 8.5 moles of sialic acid per mole of rhEPO produced by EC2-1H9-miUGCG cells and only 6 moles of sialic acid per mole of rhEPO produced by EC2-1H9 cells (Fig. 5C). This suggests that the use of EC2-1H9-miUGCG cells may represent a novel method for generating large amounts of highly sialylated rhEPO.

EC2-1H9-miUGCG cells produce rhEPO with increased levels of sialylation. (A) EPO production was determined for 4 days of culture. Closed circles, EC2-1H9 cells; Open circles, EC2-1H9-miUGCG cells. (B) Intracellular CMP-Neu5Ac levels were determined by HPLC. (C) rhEPO was purified using an immunoaffinity column with an anti-EPO antibody. Sialylation was determined using MAL I lectin. (D) Molar ratio of sialic acid on rhEPO. n = 3. **P < 0.01 (Two-tailed Student’s t-test); NS, not significant. Data are presented as means ± S.E.M.
Analysis of N-linked glycan sialylation on rhEPO
To determine how stable depletion of UCGC affects the structure of the glycosylation chains on rhEPO, we used HPLC to analyze the N-linked glycans released from rhEPO after 2-aminobezaminde (2-AB) labeling27. We used an anion-exchange column (DEAE-5PW) to separate the 2AB-labeled N-linked glycans by their number of sialic acids and then estimated rhEPO sialylation by measuring the resulting peak area. For this sialylation profiling analysis, we used the 2-AB-labeled N-glycans of bovine fetuin as a sialylation standard (Fig. 6A). Consistent with the results described above (Fig. 5C and D), this technique confirmed that the rhEPO produced by EC2-1H9-miUGCG cells is more highly sialylated than that produced by EC2-1H9 cells (Fig. 6B). Specifically, we found EC2-1H9-miUGCG cells produce rhEPO with less neutral (6.9% vs. 12.4%) and mono-sialylated (14.4% vs. 21.0%) N-glycans compared to the control rhEPO produced by EC2-1H9 cells (Fig. 6C). Instead, the rhEPO produced by EC2-1H9-miUGCG cells shows significantly more di-sialylated N-glycans (40.8% vs. 30.7%) than the control rhEPO. The rhEPO produced by EC2-1H9-miUGCG cells shows similar levels of tri-sialylated (25.0% vs. 23.8%) and tetra-sialylated (12.9% vs. 12.1%) N-glycans compared to the rhEPO produced by EC2-1H9 cells (Fig. 6C).

Analysis of sialylated N-linked glycans on rhEPO. (A) N-glycans released from rhEPO were labeled with 2-AB. 2-AB-labeled N-glycans were separated using a DEAE column by HPLC. 2-AB-labeled N-glycans of fetuin (top), rhEPO produced by EC2-1H9 cells (middle), and rhEPO produced by EC2-1H9-miUGCG cells (bottom). (B) Sialylated rhEPO determined in A. (C) Neutral, mono-, di-, tri-, and tetra-sialylated N-glycans of rhEPO quantified in A. n = 3. **P < 0.01 (Two-tailed Student’s t-test). Data are presented as means ± S.E.M.
Discussion
Sialic acid is the terminal sugar on the glycosylation chains of most secreted glycoproteins, where it contributes to their function and stability1,28,29. In exploring the increased stability of recombinant therapeutic glycoproteins by regulating sialylation, we noted that it is also required for GSL biosynthesis16. We, therefore, hypothesized that inhibition of GSL biosynthesis may enhance the sialylation of rhEPO. First, we treated cells expressing rhEPO with a GSL biosynthesis inhibitor and confirmed the expected increase in rhEPO sialylation. Ultimately, this led us to establish the stable CHO cell line, EC2-1H9-miUGCG, which produces highly sialylated rhEPO owing to its reduced expression of UGCG. We recently discovered that we were able to increase intracellular CMP-Neu5Ac levels by co-expressing the CMP-Neu5Ac transporter CST with GNE (R263L/R266Q), bypassing any negative feedback inhibition15. Surprisingly, EC2-1H9-miUGCG cells show normal levels of CMP-Neu5Ac in the cytoplasm and in the Golgi (Fig. S3). This suggests that the increased intracellular CMP-Neu5Ac caused by inhibiting GSL biosynthesis is rapidly consumed in the sialylation of rhEPO, inducing a dynamic equilibrium.
Although we do not yet fully understand the intricacies of intracellular CMP-Neu5Ac dynamics, we used various approaches to confirm that EC2-1H9-miUGCG cells produce highly sialylated rhEPO. We found that the rhEPO produced by EC2-1H9-miUGCG cells has 10.2% more di-sialylated N-glycans than that produced by EC2-1H9 cells, but less neutral and mono-sialylated N-glycans. Although we were previously able to attribute the enhanced sialylation of the rhEPO produced by EC2-1H9-CTSTrEKm cells to tetra-sialylated N-glycans15, the rhEPO produced by EC2-1H9-miUGCG cells does not show any changes in the levels of tri- and tetra-sialylated N-glycans. This means it may be possible to improve the sialylation of rhEPO produced by EC2-1H9-CTSTrEKm cells even further via UGCG depletion. In addition, because the rate at which proteins transit the Golgi complex affects glycosylation30, it may also be possible to enhance rhEPO sialylation by slowing its passage through the Golgi.
In this study, we have determined that glycoprotein sialylation can be enhanced via the inhibition of GSL biosynthesis. We extended this finding by establishing a stable CHO cell line that produces highly sialylated rhEPO by virtue of its reduced expression of the GSL biosynthesis enzyme UGCG. In summary, we have developed a novel approach that improves the efficacy of rhEPO, a therapeutically important recombinant glycoprotein, by modulating the metabolic flux of intracellular CMP-Neu5Ac.
Methods
Inhibition of GSL biosynthesis by EtDO-P4
EtDO-P4 (D-threo-1-(3′,4′-ethylenedioxy)-phenyl-2-palmitoylamino-3-pyrrolidino-1-propanol), a nanomolar inhibitor of GSL synthesis, was provided by Dr. James A. Shayman (Department of Internal Medicine, University of Michigan, Michigan, USA). For the EtDO-P4 treatment experiments, 5 × 106 cells were cultured on 100 mm plates in MEM-α (Minimum Essential Medium Eagle-α; Gibco, Grand Island NY) supplemented with 10% dFBS (dialyzed fetal bovine serum; Gibco) for 24 hours31. Cells were washed twice with serum-free medium (CHO-S-SFM II; Gibco) before culturing them for an additional 48 hours in serum-free medium containing 1 µM EtDO-P4.
Western blotting
rhEPO containing culture media was subjected to SDS-PAGE, followed by transferring to a PVDF membrane (Millipore Corp., Bedford, MA). After blocking with 5% BSA in TBS-T (140 mM NaCl, 10 mM Tris-HCl, with 0.05% Tween 20, pH 8.0) for 1 hour at room temperature, the PVDF membrane was incubated with biotinylated lectins (Wheat Germ Agglutinin; WGA, Maackia Amurensis leukoagglutinin I; MAL I) at 4 °C for 16 hours. After rinsing three times for 5 min using TBS-T, the membrane was incubated with ExtrAvidin-peroxidase (Sigma) at room temperature for 1 hour, followed by developing using ECL kit (Thermo Scientific; Rockford, IL). For reblotting, the membrane was stripped at room temperature for 1 hour with stripping buffer (Candor Bioscience GmbH, Weissensberg, Germany). PVDF membrane was rinsed 5 times with T-TBS. The membrane was blocked using 5% BSA in TBS-T, and then incubated with 1:1000 diluted anti-EPO antibody (Santa Cruz, CA). After washing three times with TBS-T for 5 min, the membrane was incubated with 1:5000 diluted HRP-labeled anti-mouse IgG antibody (Santa Cruz) at room temperature for 1 hour. The membrane was developed using ECL solution (Thermo Scientific; Rockford, IL).
Depletion of UGCG expression using siRNA and miRNA
Three siRNA sequences (siRNA1, siRNA2 and siRNA3) which targets Chinese hamster UGCG mRNAs (GenBank accession no. NM_001246692) were obtained from Invitrogen (Carlsbad, CA). The oligonucleotides are listed in Table SI. 10 μM of each UGCG-specific siRNA was transfected to the EC2-1H9 cells using Lipofectamine™ RNAiMAX reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instruction. To establish stable cell line which prevents the expression of UGCG, miRNA expression vector was prepared according to the sequence of siRNA1 (see Table S1). In brief, paired oligonucleotides were annealed in buffer (30 mM HEPES-KOH, pH 7.4, 150 mM KCl, 2 mM MgCl2) at 90 °C for 1 min followed by cooling down. To prepare the pcUGCG-miR-1 vector, DNA fragment was inserted into the pcDNATM 6.2-GW/EmGFP-miR vector (Invitrogen, Carlsbad, CA), which contains a blasticidin gene for the selection.
Total RNAs extracted from CHO cells with TRIzol® (Invitrogen, Carlsbad, CA) were used to prepare cDNA by which extracted RNAs were reversed transcribed into using AccuPower RT-PCR PreMix (Bioneer) according to the manufacturer’s instructions. Expression level of UGCG was examined by RT-PCR using following primers shown 5′ to 3′: forward (CTC GAG ATG GCG CTG CT); reverse (TCT AGA TTA TAC ATC TAG GAT TTC CTC TGC). The amplified cDNAs were separated on 0.8% agarose gel which was visualized by ethidium bromide (EtBr) staining.
Establishment of UGCG depleted CHO cell line
EC2-1H9 cells, which are CHO cells that produce recombinant human EPO, were provided by Dr. Hyo Jeong Hong (Antibody Engineering Research Unit, Korea Research Institute of Bioscience and Biotechnology, Yuseong-gu, Daejeon, Korea). Cells were maintained in MEM-α (Gibco) supplemented with 10% (v/v) dFBS (Gibco), 3.5 g/L glucose, 20 nM MTX (methotrexate; Sigma), and 1% (v/v) Ab-Am (antibiotic-antimycotic solution; Gibco) in a humidified atmosphere containing 5% CO2 at 37 °C. The pcUGCG-miR-1 was transfected into EC2-1H9 cells using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA). Transfected cells were selected by blasticidin (Sigma) for 2 weeks.
GSL extraction, thin-layer chromatography (TLC), and immunostaining
GSL extraction, TLC analysis, and immunostaining experiments were performed as previously described32. Briefly, GSLs were extracted from confluent CHO cells using chloroform/methanol (C/M; 2:1, v/v) and isopropanol/hexane/water (IPA/H/W; 55:25:20, v/v/v) under sonication for 30 min. GSL extracts were dissolved in 0.1 M NaOH/MeOH and incubated at 40 °C for 2 hours, followed by neutralization using 1 N HCl. Hexane was added and incubated at room temperature for 5 min. The lower layer was evaporated under N2 stream and resuspended using distilled water. The solution was subjected to SepPak C18 cartridge (Varian, Palo Alto, CA). After rinsing the column using distilled water, total GSLs were eluted using C/M. Total GSLs were determined on TLC (Silica Gel 60 F-254, Merck, Whitehouse Station, NJ) in C/M/0.2% CaCl2 (55:40:10, v/v/v), followed by staining using 2% orcinol in 2 M H2SO4.
Determination of the enzymatic activity of UGCG
Enzymatic activity of UGCG was determined as previously established33,34. In brief, cells (4 × 107) in TBS containing protease inhibitor (Roche, Basel, Switzerland) were lysed using sonication. Cell lysate (200 µg) was incubated in reaction buffer [50 mM Tris-HCl pH 7.5, 1 mM EDTA, 50 pmol of C6-NBD-Cer (ThermoFisher Scientific, Waltham, MA), 500 µM of UDP-Glc, 6.5 nmol of lecithin (Wako, Osaka, Japan)] at 37 °C for 60 min. Lipids were extracted by Folch method using 200 µl of chloroform/methanol (2:1, v/v) and 5 µl of 500 µM KCl. After centrifugation, the organic phase was dried using centrifugal concentrator. Samples were suspended using 15 µl of chloroform/methanol (2:1, v/v) and subjected to the TLC (Silica Gel 60, Merck) which was developed with chloroform/methanol/water (65:25:4, v/v/v). Lipids were determined by LED transilluminator at 470 nm. C6-NBD-Cer, C6-NBD-GlcCer, C6-NBD-SM were quantified using a Shimadzu CS-9300 chromatoscanner (excitation: 475 nm, emission: 525 nm).
Isolation of the Golgi complex
Golgi membrane was extracted from CHO cells as previously described35. Briefly, 1.0 × 108 CHO cells were resuspended with homogenizing buffer (0.25 M sucrose, 10 mM Tris-HCl, pH 7.4) and homogenized using ball-bearing homogenizer with 22 µm ball. The solution was subjected to sucrose gradient centrifugation at 110,000 x g for 2.5 hours. Golgi membrane was collected at the 29%/35% sucrose interface using a syringe with 18G gauge needle, and determined the protein concentration using the Quant-iT™ protein assay kit (Invitrogen).
Determination of intracellular CMP-sialic acid levels in CHO cells
The concentration of intracellular CMP-Neu5Ac in CHO cells was examined as previously described36. Briefly, 1.0 × 106 cells were lysed in cold 75% (v/v) ethanol using a cell disruptor. The soluble fraction was separated using centrifugation at 13,000 rpm at 4 °C for 10 min. The lyophilized CMP-Neu5Ac was dissolved with 120 μl of 40 mM phosphate buffer (pH 9.2), followed by centrifugation. The supernatant was filtered using a centricon (MWCO, 10,000) and subjected to CarboPac PA-1 column (Dionex, Sunnyvale, CA). CMP-sialic acid was determined by Abs260 absorbance detector (model 486; Waters). The concentration of intracellular CMP-Neu5Ac was normalized to cell number.
Production of rhEPO and determination of its sialic acid level
5.0 × 106 of EC2-1H9 cells or the engineered cells were seeded in T175 culture flask containing MEM-α supplemented with 10% (v/v) dFBS, 3.5 g/L glucose, 1% (v/v) Ab-Am solution, and 20 nM MTX. The culture medium was replaced with serum-free medium (CHO-S-SFM II; Gibco) in 3 days. After 2 days, the culture medium was collected, filtered using 0.45 μm filter (Sartorius, Göttingen, Germany), and dialyzed against phosphate buffer saline (PBS, pH 7.4) at 4 °C, overnight. To purify rhEPO, cultured medium was subjected to EPO Purification Gel (MAIIA Diagnostics, Uppsala, Sweden) according to the manufacturer’s instructions. Purified rhEPO was dialyzed against distilled water at 4 °C, overnight. The concentration was measured using a Quant-iT™ protein assay kit (Invitrogen) and stored at −80 °C until use.
The sialic acid level of the purified rhEPO was examined using the OPD-labeling method as previously described26. Briefly, sialic acid moieties from the purified rhEPO were released using 0.5 M NaHSO4 at 80 °C for 20 min. Released sialic acid was labeled with OPD (o-phenylenediamine-2HCl; Sigma) at 80 °C for 40 min. The level of OPD-labeled sialic acid was determined using C18 reversed-phase column (Shim-pack CLC-ODS; Shimadzu, Kyoto, Japan) with 474 scanning fluorescence detector (excitation at 230 nm and emission at 420 nm, Waters).
Profiling the sialylation of rhEPO’s N-linked glycans
The purified rhEPO (50 μg) was denatured by heating at 95 °C for 5 min. N-glycans were released from rhEPO by incubation with PNGase F (Promega) at 37 °C for 3 hours and purified using GlycoClean R cartridges (Glyko; ProZyme, Hayward, CA). Purified N-glycans were labeled with 2-AB using 2-AB labeling kit (Glyko; ProZyme, Hayward, CA). The sample was subjected to an anion-exchange column (TSKgel DEAE-5PW, 7.5 mm × 75 mm; Tosoh, Tokyo, Japan) as described previously (Llop et al. 2007). These included solvent A (50% 0.5 M ammonium formate (pH 4.5)/acetonitrile (ACN)/distilled water (5:2:3, v/v/v)) and solvent B (20% (v/v) ACN in distilled water). 2AB-labeled N- glycans were separated in a linear gradient of buffer B from 0% (v/v) to 100% (v/v) over 45 min at a flow rate of 0.4 mL/min at 30 °C. 2-AB N-glycans of bovine fetuin (Glyko) were used as a standard.
References
Ngantung, F. A., Miller, P. G., Brushett, F. R., Tang, G. L. & Wang, D. I. C. RNA interference of sialidase improves glycoprotein sialic acid content consistency. Biotechnology and Bioengineering 95, 106–119 (2006).
Fukuda, M., Sasaki, H., Lopez, L. & Fukuda, M. Survival of recombinant erythropoietin in the circulation: the role of carbohydrates. Blood 73, 84–89 (1989).
Weiss, P. & Ashwell, G. The asialoglycoprotein receptor: properties and modulation by ligand. Progress in Clinical and Biological Research 300, 169–184 (1989).
Weikert, S. et al. Engineering Chinese hamster ovary cells to maximize sialic acid content of recombinant glycoproteins. Nat Biotech 17, 1116–1121 (1999).
Fukuta, K., Yokomatsu, T., Abe, R., Asanagi, M. & Makino, T. Genetic engineering of CHO cells producing human interferon-γ by transfection of sialyltransferases. Glycoconjugate Journal 17, 895–904 (2000).
Jeong, Y. T. et al. Enhanced sialylation of recombinant erythropoietin in CHO cells by human glycosyltransferase expression. Journal of Microbiology and Biotechnology 18, 1945–1952 (2008).
Kornfeld, S., Kornfeld, R., Neufeld, E. F. & O’Brien, P. J. The feedback control of sugar nucleotide biosynthesis in liver. Proceedings of the National Academy of Sciences of the United States of America 52, 371–379 (1964).
Kean, E. L. Nuclear Cytidine 5′-Monophosphosialic Acid Synthetase. Journal of Biological Chemistry 245, 2301–2308 (1970).
Yan, J.-P. et al. 3′-Azidothymidine (Zidovudine) Inhibits Glycosylation and Dramatically Alters Glycosphingolipid Synthesis in Whole Cells at Clinically Relevant Concentrations. Journal of Biological Chemistry 270, 22836–22841 (1995).
Gu, X. & Wang, D. I. C. Improvement of interferon-γ sialylation in Chinese hamster ovary cell culture by feeding of N-acetylmannosamine. Biotechnology and Bioengineering 58, 642–648, (1998).
Baker, K. N. et al. Metabolic control of recombinant protein N-glycan processing in NS0 and CHO cells. Biotechnology and Bioengineering 73, 188–202 (2001).
Wong, N. S. C., Yap, M. G. S. & Wang, D. I. C. Enhancing recombinant glycoprotein sialylation through CMP-sialic acid transporter over expression in Chinese hamster ovary cells. Biotechnology and Bioengineering 93, 1005–1016 (2006).
Viswanathan, K., Narang, S., Hinderlich, S., Lee, Y. C. & Betenbaugh, M. J. Engineering Intracellular CMP-Sialic Acid Metabolism into Insect Cells and Methods to Enhance Its Generation. Biochemistry 44, 7526–7534 (2005).
Bork, K., Reutter, W., Weidemann, W. & Horstkorte, R. Enhanced sialylation of EPO by overexpression of UDP-GlcNAc 2-epimerase/ManAc kinase containing a sialuria mutation in CHO cells. FEBS Letters 581, 4195–4198 (2007).
Son, Y.-D., Jeong, Y. T., Park, S.-Y. & Kim, J. H. Enhanced sialylation of recombinant human erythropoietin in Chinese hamster ovary cells by combinatorial engineering of selected genes. Glycobiology 21, 1019–1028 (2011).
Hakomori, S.-i. The glycosynapse. Proceedings of the National Academy of Sciences 99, 225–232 (2002).
Colley, K. J., Lee, E. U., Adler, B., Browne, J. K. & Paulson, J. C. Conversion of a Golgi apparatus sialyltransferase to a secretory protein by replacement of the NH2-terminal signal anchor with a signal peptide. Journal of Biological Chemistry 264, 17619–17622 (1989).
Deutscher, S. L., Nuwayhid, N., Stanley, P., Briles, E. I. B. & Hirschberg, C. B. Translocation across golgi vesicle membranes: A CHO glycosylation mutant deficient in CMP-sialic acid transport. Cell 39, 295–299 (1984).
Lee, L., Abe, A. & Shayman, J. A. Improved Inhibitors of Glucosylceramide Synthase. Journal of Biological Chemistry 274, 14662–14669 (1999).
Fedoryszak-Kuśka, N., Panasiewicz, M., Domek, H. & Pacuszka, T. Glucosylceramide synthase inhibitors D-PDMP and D-EtDO-P4 decrease the GM3 ganglioside level, differ in their effects on insulin receptor autophosphorylation but increase Akt1 kinase phosphorylation in human hepatoma HepG2 cells. Acta Biochimica Polonica 63, 247–251 (2016).
Park, S.-Y., Yoon, S.-J., Freire-de-Lima, L., Kim, J.-H. & Hakomori, S.-I. Control of cell motility by interaction of gangliosides, tetraspanins, and epidermal growth factor receptor in A431 versus KB epidermoid tumor cells. Carbohydrate Research 344, 1479–1486 (2009).
Yoon, S.-J., Nakayama, K.-I., Hikita, T., Handa, K. & Hakomori, S.-I. Epidermal growth factor receptor tyrosine kinase is modulated by GM3 interaction with N-linked GlcNAc termini of the receptor. Proceedings of the National Academy of Sciences of the United States of America 103, 18987–18991 (2006).
Jeong, Y. T., Choi, O., Son, Young, D., Park, Seung, Y. & Kim, J. H. Enhanced sialylation of recombinant erythropoietin in genetically engineered Chinese-hamster ovary cells. Biotechnology and Applied Biochemistry 52, 283–291 (2009).
Monsigny, M., Roche, A.-C., Sene, C., Maget-Dana, R. & Delmotte, F. Sugar-Lectin Interactions: How Does Wheat-Germ Agglutinin Bind Sialoglycoconjugates? European Journal of Biochemistry 104, 147–153 (1980).
Takeuchi, M. et al. Comparative study of the asparagine-linked sugar chains of human erythropoietins purified from urine and the culture medium of recombinant Chinese hamster ovary cells. Journal of Biological Chemistry 263, 3657–3663 (1988).
Anumula, K. R. Rapid Quantitative Determination of Sialic Acids in Glycoproteins by High-Performance Liquid Chromatography with a Sensitive Fluorescence Detection. Analytical Biochemistry 230, 24–30 (1995).
Llop, E. et al. Evaluation of protein N-glycosylation in 2-DE: Erythropoietin as a study case. PROTEOMICS 7, 4278–4291 (2007).
Varki, A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 3, 97–130 (1993).
Arnold, J. N., Wormald, M. R., Sim, R. B., Rudd, P. M. & Dwek, R. A. The Impact of Glycosylation on the Biological Function and Structure of Human Immunoglobulins. Annual Review of Immunology 25, 21–50 (2007).
Reynders, E., Foulquier, F., Annaert, W. & Matthijs, G. How Golgi glycosylation meets and needs trafficking: the case of the COG complex. Glycobiology 21, 853–863 (2011).
Park, S.-Y., Kwak, C.-Y., Shayman, J. A. & Kim, J. H. Globoside promotes activation of ERK by interaction with the epidermal growth factor receptor. Biochimica et Biophysica Acta (BBA) - General Subjects 1820, 1141–1148 (2012).
Kannagi, R., Nudelman, E., Levery, S. B. & Hakomori, S. A series of human erythrocyte glycosphingolipids reacting to the monoclonal antibody directed to a developmentally regulated antigen SSEA-1. Journal of Biological Chemistry 257, 14865–14874 (1982).
Hayashi, Y., Horibata, Y., Sakaguchi, K., Okino, N. & Ito, M. A sensitive and reproducible assay to measure the activity of glucosylceramide synthase and lactosylceramide synthase using HPLC and fluorescent substrates. Analytical Biochemistry 345, 181–186 (2005).
Ichikawa, S., Sakiyama, H., Suzuki, G., Hidari, K. I. & Hirabayashi, Y. Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proceedings of the National Academy of Sciences 93, 4638–4643 (1996).
Park, S.-Y., Yang, J.-S. & Hsu, V. W. In The Golgi Complex: Methods and Protocols (ed William J. Brown) 63–74 (Springer New York, 2016).
Tomiya, N., Ailor, E., Lawrence, S. M., Betenbaugh, M. J. & Lee, Y. C. Determination of Nucleotides and Sugar Nucleotides Involved in Protein Glycosylation by High-Performance Anion-Exchange Chromatography: Sugar Nucleotide Contents in Cultured Insect Cells and Mammalian Cells. Analytical Biochemistry 293, 129–137 (2001).
Acknowledgements
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) & funded by the Korean government (MSIP&MOHW) (No. 2013M3A9B6075931).
Author information
Authors and Affiliations
Contributions
C.Y.K., S.Y.P., C.G.L., and N.O. performed the experiments. All authors participated in the experimental design and data analysis. C.Y.P., S.Y.P., N.O., M.I., and J.H.K. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kwak, CY., Park, SY., Lee, CG. et al. Enhancing the sialylation of recombinant EPO produced in CHO cells via the inhibition of glycosphingolipid biosynthesis. Sci Rep 7, 13059 (2017). https://doi.org/10.1038/s41598-017-13609-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13609-4
This article is cited by
-
Structural basis for the delivery of activated sialic acid into Golgi for sialyation
Nature Structural & Molecular Biology (2019)
-
Inhibition of poly-LacNAc biosynthesis with release of CMP-Neu5Ac feedback inhibition increases the sialylation of recombinant EPO produced in CHO cells
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
