
Abstract
Phyllomeroterpenoids A−C (1−3), multi-biosynthetic pathway derived meroterpenoids from amino acid/pentose phosphate/terpenoid pathways, were isolated from the TCM endophytic fungus Phyllosticta sp. J13-2-12Y, together with six biosynthetically related compounds (4−9). All structures were determined by extensive spectroscopic analysis, chemical derivatization, and ECD experiments. A plausible biosynthetic pathway of 1−3 was proposed. In addition, the antimicrobial activities of all isolated compounds were evaluated against Staphylococcus aureus 209P (bacterium) and Candida albicans FIM709 (fungus).
Similar content being viewed by others

Introduction
Meroterpenoids, such as fumagillin1, mycophenolic acid2, avinosol3, merochlorin A4, cochlearol B5, and others, have received much attention from chemists and pharmacologists6 for their remarkable structural diversity and varied biological activities. In general, meroterpenoids originate from a dual-biosynthetic pathway6,7. This is composed of a non-terpenoid pathway and a terpenoid pathway, such as the polyketide/terpenoid and shikimate/terpenoid pathways.
Acorus tatarinowii is a common and important medicinal plant, and its dried rhizomes have a long history of being used as traditional Chinese medicine (TCM) as Shi Chang Pu (Acori Tatarinowii Rhizoma) to treat many diseases, such as nervous ailments, dysentery, bronchitis, intermittent fevers ect8. A. tatarinowii is rich with asarones, which show antimicrobial activity8,9. The micro-environment of A. tatarinowii is special due to the existence of abundant antimicrobial asarones, and the microorganisms living in this habitat should be distinctive. During our recent search for bioactive compounds from microorganisms10,11,12,13,14, chemical investigation on a TCM endophytic fungal strain of Phyllosticta sp. J13-2-12Y from the leaves of A. tatarinowii was carried out. Through this investigation, three unusual meroterpenoids, phyllomeroterpenoids A–C (1−3), were isolated, along with six biosynthetically related compounds (4−9) (Fig. 1). Phyllomeroterpenoids A–C (1−3) are multi-biosynthetic pathway derived meroterpenoids, whose structures are composed of one guignardianone unit from the amino acid pathway, one C7 unit from the pentose phosphate pathway, and one monoterpene unit from the terpenoid pathway. The guignardianone unit is the skeleton of the guignardianone derivative, while the C7 unit and the monoterpene unit compose the guignardone-type meroterpenoids. In this study, we report the isolation and structural elucidation of 1−9 as well as their antimicrobial activities. In addition, a plausible biogenetic pathway of 1−3 is proposed.

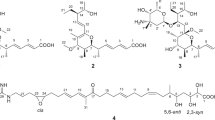
Chemical structures of 1−9.
Results
The known compounds, including three guignardianones (4−6) and three guignardone-type meroterpenoids (7−9) were identified as (S, Z)-guignardianone C (4)15,16, (S, Z)-botryosphaerin B (5)17, (S, Z)-phenguignardic acid methyl ester (6)15,16, (4S, 6R, 9S, 10R, 14R)−17-hydroxylated guignardone A (7)18, (4S, 6R, 9S, 10R, 14R)-guignardone B (8)19, and (4S, 6R, 9S, 10S, 12S, 14R)−12-hydroxylated guignardone A (9)18 by comparisons of their NMR (recorded in CDCl3) and ECD data with references. In addition, the NMR data of 4 in CD3OD (Tables S4), 8 in CD3OD (Tables S5), and 9 in DMSO-d 6 (Tables S6) are reported for the first time.
Phyllomeroterpenoid A (1) was obtained as a yellowish oil. The positive ion at m/z 551.2277 [M + H]+ (calcd. for C31H35O9, 551.2281) from HRESIMS indicated the molecular formula of C31H34O9 (index of hydrogen deficiency = 15). In the1H NMR spectrum of 1, the characteristic signals of five aromatic protons [δ H 7.67 (2H), 7.41 (2H), 7.35 (1H)], three olefinic protons [δ H 6.50 (1H, s), 5.11 (1H, br s), 4.91 (1H, br s)], one O-methine [δ H 4.54 (1H, d, J = 5.5 Hz)], two O-methylenes [δ H 4.64 (2H, br s), 3.79 (1H, d, J = 7.9 Hz), 3.47 (1H, d, J = 7.9 Hz)], and three methyls [δ H 1.29 (3H, s), 1.07 (6H, d, J = 6.9 Hz)] were observed. Among them, the five aromatic protons indicated the existence of a mono-substituted benzene ring moiety in 1. Combined with the DEPT-135 spectrum, 31 signals were observed in the13C NMR spectrum, which can be assigned to eight sp2 quaternary carbons (including one ketone carbonyl and two ester carbons), six sp2 methine carbons, one sp2 methylene carbon, three sp3 O-quaternary carbons, four sp3 methine carbons (including one O-methine carbon), six sp3 methylene carbons (including two O-methylene carbons), and three methyl carbons. Based on the analysis of1H−1H COSY experiment, four subunits (C-4—C-5, C-8—C-9—C-14—C-13—C-12, C-5′—C-6′—C-7′—C-8′—C-9′, and C-13′—C-12′—C-14′) were revealed as shown in Fig. 2. Combined with the analysis of1H−1H COSY, the HMBC correlations (Fig. 2) from H-4 to C-2/C-3/C-6/C-7, from Ha-5/Hb-5 to C-1/C-3/C-6/C-7, from Ha-7/Hb-7 to C-1/C-4/C-5/C-6, from Ha-8/Hb-8 to C-1/C-2/C-3/C-10/C-14, from H-9 to C-10, from H3-11 to C-9/C-10/C-12, from Ha-16/Hb-16 to C-14/C-15/C-17, and from H2-17 to C-14/C-15/C-16 revealed a guignardone-type meroterpenoid moiety in 1. In addition, the HMBC correlations (Fig. 2) from H-3′ to C-1′/C-2′/C-5′/C-9′, from H-5′/H-9′ to C-3′, from H-6′/H-8′ to C-4′, from H-7′ to C-5′/C-9′, from H-12′ to C-10′/C-11′, from H3-13′ to C-11′/C-12′/C-14′, and from H3-14′ to C-11′/C-12′/C-13′ revealed a guignardianone moiety in 1, combined with a comparison of NMR data with (S, Z)-guignardianone C (4)15,16 and the above analysis of1H−1H COSY. Based on the molecular formula and the key HMBC correlation from H2-17 to C-10′, these two moieties can be combined, and the planar structure was established as shown in Fig. 2. This is the ester of a guignardone-type meroterpenoid and a guignardianone, and the assignments of NMR data can be found in Table 1.

Key1H-1H COSY, HMBC, and NOESY correlations of 1.
The key NOESY correlations (Table S1) between H-5′/H-9′ and H3-13′/H3-14′ indicated that the configuration of the double bond of ∆2′ as Z. Furthermore, the13C NMR data of guignardianone unit in 1 were quite similar to those of (S, Z)-guignardianone C (4)14,16, which confirmed the above deduction. In addition, the alkaline hydrolysis of 1 give a major reaction product (1a) that was identified as (4S, 6R, 9S, 10R, 14R)−17-hydroxylated guignardone A (7)18 by HPLC, NMR data, and electronic circular dichroism (ECD) comparisons (Figures S1–S3). Thus, the absolute configurations of C-4, C-6, C-9, C-10, and C-14 in 1 were deduced to be the same as those in 7.
The structure of 1 is composed of a guignardone moiety and a guignardianone moiety, so the observed ECD of 1 should result from the summed contributions of these two moieties based on the ECD additivity rule in diketones19. According to the structure, 7 can represent the contribution of the guignardone moiety, while 4 or the enantiomer of 4 can represent the contribution of the guignardianone moiety. The simulated ECD curve of 1, which was the sum of the experimental ECD data of 4 and 7, was similar to that of the experimental ECD curve of 1 (Fig. 3), therefore we deduced that the configuration of C-11′ in 1 should be the same as that in 4. Thus, the absolute configuration of 1 was determined as 4S, 6R, 9S, 10R, 14R, 11′S.

The experimental ECD spectra of 1, 4, and 7 and the simulated ECD spectrum of 1 (the sum of 4 and 7).
Phyllomeroterpenoid B (2) was obtained as a yellowish oil. The positive ion at m/z 553.2455 [M + H]+ (calcd. for C31H37O9, 553.2438) from HRESIMS indicated the molecular formula of C31H36O9 (index of hydrogen deficiency = 14). The detailed 2D NMR analysis (Table S2) and the comparison of NMR data with (S, Z)-guignardianone C (4) (Table S4) revealed that 2 was the ester of a guignardone-type meroterpenoid and a guignardianone unit. A precise comparison of 1D NMR data of 2 (Table 1) with (4S, 6R, 9S, 10R, 14R)-guignardone B (8) (Table S5) showed an obviously downfield shifted carbon at C-15, which suggested that the esterification was at C-15 in 2. Therefore, the planar structure of 2 was established as shown in Fig. 1. Combined with the carbon NMR data comparison with (S, Z)-guignardianone C (4) (Table S4), the key NOESY correlations (Table S2) between H-5′/H-9′ and H3-13′/H3-14′ revealed the configuration of the double bond of ∆2′ as Z. With the same alkaline hydrolysis experiment (Figures S4–S6) and the comparison analysis of the simulated ECD with the experimental ECD data (Fig. 4) as described in 1, the absolute configuration of 2 was determined as 4S, 6R, 9S, 10R, 14R, 11′S.

The experimental ECD spectra of 2, 4, and 8 and the simulated ECD spectrum of 2 (the sum of 4 and 8).
Phyllomeroterpenoid C (3) was obtained as a yellowish oil, and its molecular formula was the same as that of 1 (C31H34O9) as determined by HRESIMS. Based on comparison of the NMR data with (S, Z)-guignardianone C (4)15,16 and detailed NMR analyses (Table S3), the planar structure of 3 was established as shown in Fig. 1, and the assignments of NMR data can be found in Table 1.
Combined with the carbon NMR data comparison with (S, Z)-guignardianone C (4)15,16, the key NOESY correlations (Fig. 5) between H-5′/H-9′ and H3-13′/H3-14′ indicated the configuration of the double bond of ∆2′ as Z. In addition, the key NOESY correlations (Fig. 5) between H3-11 and H-9/Hb-8, between Hb-13 and H-9, between Ha-8 and H-14, between H-12 and H-14, and between H-14 and Hb-7, and the coupling constants of3 J H-12, Hb-13 (2.2 Hz) and3 J H-12, Ha-13 (6.9 Hz) in 3 were the same as those in 9 (Table S6), indicating that the relative configuration of the guignardone moiety in 3 is 4S*, 6R*, 9S*, 10S*, 12S*, and 14R*, which is the same as 9. Since 3 and 9 coexist in Phyllosticta sp. J13-2-12Y, the configurations of C-4, C-6, C-9, C-10, C-12, and C-14 in 3 should be the same as those in 9.

Key NOESY correlations of 3.
With the same comparison analysis of the simulated ECD with the experimental ECD data (Fig. 6) as described in 1, the absolute configuration of 3 was determined as 4S, 6R, 9S, 10S, 12S, 14R, 11′S.

The experimental ECD spectra of 3, 4, and 9 and the simulated ECD spectrum of 3 (the sum of 4 and 9).
The antimicrobial activities of the isolated compounds were evaluated against Staphylococcus aureus 209P (bacterium) and Candida albicans FIM709 (fungus). All compounds exhibited different antimicrobial activities (Table 2). Especially, 6 displayed obvious antimicrobial activities against S. aureus 209P and C. albicans FIM709 with MIC values of 4 μg/mL.
Discussion
Guignardone-type meroterpenoids are a rare kind of meroterpenoids that are composed of one C7 unit and one monoterpene unit derived from the pentose phosphate/terpenoid pathways20. Up to present, about 30 members18,21,22,23,24,25,26,27,28 with tricyclic or tetracyclic skeletons have been reported from Guignardia sp.18,21,22,23,24,25,26, Pycnoporus sanguineus
27, and Aspergillus sp.28, and they showed antifungal18, antibacterial23, cytotoxic25, and Toll-Like Receptor 3 regulating activities26. Guignardianones are a special kind of fungal-derived benzylidene dioxolanones derived from the amino acid pathway15,29,30, and they exhibit antifungal18 and antibacterial31 activities. Up to now, only 13 natural guignardianones have been reported from Guignardia sp.15,16,18,28,31,32, Botryosphaeria sp.17 and Aspergillus sp.30. On the basis of our chemical investigation, three known guignardianones (4–6), and three known guignardone-type meroterpenoids (7–9) were isolated from the TCM endophytic fungal strain of Phyllosticta sp. J13-2-12Y. In addition, unusual meroterpenoids (1–3), the heterozygotes of guignardianone and guignardone-type meroterpenoid were also obtained. Phyllomeroterpenoids A–C (1–3) are composed of one guignardianone unit, one C7 unit, and one monoterpene unit, and they are multi-biosynthetic pathway derived meroterpenoids from the amino acid/pentose phosphate/terpenoid pathways. They could originate from phenyalanine30, 2-epi−5-epi-valiolone (EEV)33, and a monoterpenoid as shown in Fig. 7.

Plausible biosynthetic pathway of compounds 1−3.
Materials and Methods
General Experimental Procedures
The detail of instruments applied in this work are provided in supporting information.
Fungal Material
The strain numbered as J13-2-12Y was isolated from the leaves of A. tatarinowii collected from Guangxi Medicinal Botanical Garden, Guangxi Province, People’s Republic of China. The details of isolation and identification can be found in supporting information.
Extraction and Isolation
The fermented material was extracted with EtOAc for three times, and the solvent was evaporated to dryness under vacuum to obtain a crude extract (42.8 g). Then the crude extract was separated by silica gel CC (4 × 15 cm) with a elution system of cyclohexane-MeOH (100:0 and 0:100, v/v) to yield a cyclohexane portion (C, 24.7 g) and a MeOH portion (W, 15.7 g). The MeOH portion (W, 15.7 g) was subjected to ODS CC (4 × 30 cm) eluting with MeOH–H2O (50:50, 70:30, 85:15, and 100:0, v/v) to yield 4 fractions (W1–W4). Fraction W2 (3.2 g) was further separated by MPLC on ODS CC (4 × 45 cm) with a elution system of MeOH-H2O (30:70 to 100:0, v/v) for 420 min at 20 mL/min to afford fractions W2-1–W2-6. Fraction W2-6 (0.4 g) was separated by silica gel CC using a elution system of cyclohexane- EtOAc (100:0 to 0:100, v/v) to yield 7 fractions (W2-6-1–W2-6-7). 1 (tR:66.0 min, 5.0 mg) was isolated from fraction W2-6-2 (20.0 mg) by preparative HPLC using MeCN–H2O (55:45, v/v) at 3 mL/min. 2 (tR: 58.0 min, 4.0 mg) was obtained from fraction W2-6-4 (15.0 mg) by preparative HPLC with MeCN–H2O (55:45, v/v) at 3 mL/min. 3 (tR: 38.5 min, 1.5 mg) was separated from fraction W2-6-7 (19.0 mg) by preparative HPLC using MeCN–H2O (60:40, v/v) at 3 mL/min. 5 (tR: 36.6 min, 7.0 mg) was isolated from fraction W2-2 (0.6 g) by preparative HPLC using MeOH–H2O (50:50, v/v) at 3 mL/min. 4 (tR: 11.0 min, 270.0 mg) was isolated from fraction W2–4 (0.6 g)by preparative HPLC using MeOH–H2O (69:31, v/v) at 3 mL/min. 6 (tR: 11.0 min, 85.0 mg) was isolated from fraction W2–5 (0.4 g) by preparative HPLC using MeCN–H2O (55:45, v/v) at 3 mL/min. Fraction W1 (4.6 g) was further separated by MPLC on ODS CC (4 × 45 cm) eluted with MeOH-H2O (20:80 to 100:0, v/v) for 400 min at 20 mL/min to afford 6 fractions (W1-1–W1–6). Fraction W1-2 (0.7 g) was subjected to silica gel CC with a elution system of cyclohexane-EtOAc (100:0 to 0:100, v/v) to obtain 4 fractions (W1-2-1–W1-2-4). 8 (tR: 18.8 min, 44.0 mg) was isolated from fraction W1-2-3 (122.0 mg) by preparative HPLC using MeCN–H2O (25:75, v/v) at 3 mL/min to yield. 7 (tR: 14.7 min, 4.0 mg) and 9 (tR: 15.5 min, 5.0 mg) were isolated from fraction W1-2-2 (87.0 mg) by preparative HPLC using MeCN–H2O (28:72, v/v) at 3 mL/min.
Spectroscopic data of 1–3
Phyllomeroterpenoid A (1): yellowish oil; [α]27 D –38.3 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 204 (3.67), 222 (3.44), 269 (3.66), 295 (3.68), 308 (3.60); IR (KBr) vmax 3441, 2938, 1796, 1755, 1658, 1616, 1450, 1364, 1256, 1178, 1029, 977, 905, 758, 689 cm−1; ECD λ nm (∆ε) (c 0.9 × 10−4 mol/L, MeOH) 222 (+9.53), 255 (+12.77), 292 (–14.64) nm; ESI-MS (positive): m/z 1123 [2M + Na]+, 573 [M + Na]+; HRESIMS (positive): m/z 551.2277 [M + H]+ (calcd. for C31H35O9, 551.2281).
Phyllomeroterpenoid B (2): yellowish oil; [α]27 D –18.7 (c 0.10, MeOH). UV (MeOH) λmax (log ε) 204 (3.67), 222 (3.46), 267 (3.66), 295 (3.61), 309 (3.55). IR (KBr) vmax 3447, 2979, 2935, 1799, 1746, 1655, 1619, 1450, 1370, 1299, 1249, 1181, 1124, 1036, 977, 758, 693 cm−1; ECD λ nm (∆ε) (c 0.9 × 10−4 mol/L, MeOH) 221 (+10.02), 258 (+11.33), 295 (–9.88) nm; ESI-MS (positive): m/z 575 [M + Na]+, 553 [M + H]+; HRESIMS (positive): m/z 553.2455 [M + H]+ (calcd. for C31H37O9, 553.2438).
Phyllomeroterpenoid C (3): yellowish oil; [α]27 D –45.0 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 204 (3.82), 222 (3.62), 263 (3.93), 294 (3.69), 308 (3.62); IR (KBr) vmax 3435, 2924, 1796, 1755, 1655, 1613, 1447, 1382, 1246, 1038, 891, 684 cm−1; ECD λ nm (∆ε) (c 0.9 × 10−4 mol/L, MeOH) 221 (+14.16), 260 (+24.27), 293 (–18.84) nm; ESI-MS (positive): m/z 1123 [2M + Na]+, 573 [M + Na]+; HRESIMS (positive): m/z 551.2285 [M + H]+ (calcd. for C31H35O9, 551.2281).
Alkaline hydrolysis of 1 and 2
A sample of 1 (1 mg) was treated with 2 N KOH (200 μL), THF (200 μL), and CH3OH (200 μL), and stirred at 25 °C for 4 h. After neutralizing with 10% HCOOH and extracting with EtOAc, the EtOAc layer was evaporated to dryness and dissolved in MeOH. Then, 1a (0.4 mg) was isolated from the mixture by analytical HPLC (Phenomenex Gemini C18 column, 5 μm, 4.6 × 250 mm) with MeOH-H2O (69:31, v/v) at 1 mL/min, and its 1H NMR spectrum and ECD spectrum were identical with those of 7 (Figures S2 and S3).
A sample of 2 (1 mg) was treated with 2 N KOH (200 μL), THF (200 μL), and CH3OH (200 μL), and stirred at 25 °C for 4 h. After neutralizing with 10% HCOOH and extracting with EtOAc, the EtOAc layer was evaporated to dryness and dissolved in MeOH. Then, 2a (0.4 mg) was isolated from the mixture by analytical HPLC (Phenomenex Gemini C18 column, 5 μm, 4.6 × 250 mm) with MeOH-H2O (67:33, v/v) at 1 mL/min, and its 1H NMR spectrum and ECD spectrum were identical with those of 8 (Figures S5 and S6).
Antimicrobial Assay
The antimicrobial activities against S. aureus 209P and C. albicans FIM709 were measured in sterile 96-well plates using the broth microdilution method34,35, and the detail can be found in supporting information.
References
McCowen, M. C., Callender, M. E. & Lawlis, J. F. Fumagillin (H-3), a new antibiotic with amebicidal properties. Science 113, 202–203 (1951).
Sintchak, M. D. et al. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophenolic acid. Cell 85, 921–930 (1996).
Diaz-Marrero, A. R. et al. Avinosol, a meroterpenoid-nucleoside conjugate with antiinvasion activity isolated from the marine sponge Dysidea sp. J. Org. Lett. 8, 3749–3752 (2006).
Kaysser, L. et al. Merochlorins A-D, cyclic meroterpenoid antibiotics biosynthesized in divergent pathways with vanadium-dependent chloroperoxidases. J. Am. Chem. Soc 134, 11988–11991 (2012).
Dou, M. et al. Cochlearols A and B, polycyclic meroterpenoids from the fungus Ganoderma cochlear that have renoprotective activities. Org. Lett. 16, 93–115 (2014).
Geris, R. & Simpson, T. J. Meroterpenoids produced by fungi. Nat. Prod. Rep. 26, 1063–1094 (2009).
Matsuda, Y. & Abe, I. Biosynthesis of fungal meroterpenoids. Nat. Prod. Rep. 33, 26–53 (2016).
Feng, X. L., Yu, Y., Qin, D. P., Gao, H. & Yao, X. S. Acorus linnaeus: a review of traditional uses, phytochemistry and neuropharma-cology. RSC Adv. 5, 5173–5182 (2015).
Momin, R. A. & Nair, M. G. Pest-managing efficacy of trans-asarone isolated from Daucus carota L. seeds. J. Agric. Food Chem. 50, 4475–4478 (2002).
Wang, C. X. et al. Aldgamycins J-O, 16-membered macrolides with a branched octose unit from Streptomycetes sp. and their antibacterial activities. J. Nat. Prod. 79, 2446–2454 (2016).
Gao, Y. M. et al. Adeninealkylresorcinol, the first alkylresorcinol tethered with nucleobase from Lasiodiplodia sp. Fitoterapia 112, 254–259 (2016).
Wang, C. X. et al. Same data, different structures: diastereoisomers with substantially identical NMR data from nature. Chem. Commun. 52, 1250–1253 (2016).
Zhao, H. et al. Dimericbiscognienyne A: A meroterpenoid dimer from Biscogniauxia sp. with new skeleton and its activity. Org. Lett. 19, 38–41 (2017).
Sun, T. Y. et al. A set of interesting sequoiatones stereoisomers from a wetland soil-derived fungus Talaromyces flavus. Acta Pharm. Sin. B. 7, 167–172 (2017).
Buckel, I. et al. Phytotoxic dioxolanone-type secondary metabolites from Guignardia bidwellii. Phytochemistry 89, 96–103 (2013).
Andernach, L. et al. Assignment of configuration in a series of dioxolanone-type secondary metabolites from Guignardia bidwellii–a comparison of VCD and ECD spectroscopy. Eur. J. Org. Chem. 5946–5951 (2013).
Ju, Z. R. et al. New phenyl derivatives from endophytic fungus Botryosphaeria sp. SCSIO KcF6 derived of mangrove plant Kandelia candel. Nat. Prod. Res. 30, 192–198 (2016).
Li, T. X., Yang, M. H., Wang, X. B., Wang, Y. & Kong, L. Y. Synergistic antifungal meroterpenes and dioxolanone derivatives from the endophytic fungus Guignardia sp. J. Nat. Prod. 78, 2511–2520 (2015).
Lightner, D. A., Gurst, J. E. Organic Conformational Analysis and Sterochemistry from Circular Dichroism Spectroscopy. 306 (John Wiley & Sons: New York, 2000).
Liebermann, B., Nussbaum, R. P., Günther, W. & Teuscher, J. M. Biosynthesis of the bicycloalternarenes, mixed terpenoids of Alternaria alternate. Phytochemistry 56, 551–557 (2001).
Yuan, W. H. et al. Guignardones A–C: Three meroterpenes from Guignardia mangiferae. Eur. J. Org. Chem. 6348–6353 (2010).
Zheng, B. et al. Two new meroterpenes from endophytic fungus A1 of Scyphiphora hydrophyllacea. J. Asian. Nat. Prod. Res. 14, 776–779 (2012).
Mei, W. L. et al. Meroterpenes from endophytic fungus A1 of mangrove plant Scyphiphora hydrophyllacea. Mar. Drugs 10, 1993–2001 (2012).
Guimarães, D. O., Lopes, N. P. & Pupo, M. T. Meroterpenes isolated from the endophytic fungus Guignardia mangiferae. Phytochemistry Lett. 5, 519–523 (2012).
Sun, Z. H. et al. Guignardones P–S, new meroterpenoids from the endophytic fungus Guignardia mangiferae A348 derived from the medicinal plant smilax glabra. Molecules 20, 22900–22907 (2015).
Han, W. B. et al. Meroterpenes with toll-like receptor 3 regulating activity from the endophytic fungus Guignardia mangiferae. Planta Med. 81, 145–151 (2015).
Molinar, E. et al. Coibanoles, a new class of meroterpenoids produced by Pycnoporus sanguineus. Tetrahedron Lett. 53, 919–922 (2012).
Bai, Z. Q. et al. New meroterpenoids from the endophytic fungus Aspergillus flavipes AIL8 derived from the mangrove plant Acanthus ilicifolius. Mar. Drugs 13, 237–248 (2015).
Molitor, D. et al. Phenguignardic acid and guignardic acid, phytotoxic secondary metabolites from Guignardia bidwellii. J. Nat. Prod. 75, 1265–1269 (2012).
Bai, Z. Q. et al. New phenyl derivatives from endophytic fungus Aspergillus flavipes AIL8 derived of mangrove plant Acanthus ilicifolius. Fitoterapia 95, 194–202 (2014).
Rodrigues-Heerklotz, K. F., Heerklotz, J., Werner, C. Extracts from the fungus Guignardia sp., their uses in pharmaceutical compositions, new isolate compound from the extract of the fungus Guignardia sp. and its use in pharmaceutical compositions. EP. Patent WO 0217937, June 18 (2003).
Rodrigues-Heerklotz, K. F., Drandarov, K., Heerklotz, J., Hesse, M. & Werner, C. Guignardic acid, a novel type of secondary metabolite produced by the endophytic fungus Guignardia sp.: Isolation, structure elucidation, and asymmetric synthesis. Helv. Chim. Acta 84, 3766–3772 (2001).
Osborn, A. R. et al. Evolution and distribution of C7−cyclitol synthases in prokaryotes and eukaryotes. ACS Chem. Biol. 12, 979–988 (2017).
Groblacher, B., Maier, V., Kunert, O. & Bucar, F. Putative mycobacterial efflux inhibitors from the seeds of Aframomum melegueta. J. Nat. Prod. 75, 1393–1399 (2012).
Qin, X. J. et al. Antibacterial prenylbenzoic acid derivatives from Anodendron formicinum. Fitoterapia 92, 238–243 (2014).
Acknowledgements
This work was financially supported by grants from the National Natural Science Foundation of China (81422054 and 81373306), the Guangdong Natural Science Funds for Distinguished Young Scholar (S2013050014287), Guangdong Special Support Program (2016TX03R280), Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (Hao Gao, 2014), Pearl River Nova Program of Guangzhou (201610010021), K. C. Wong Education Foundation (Hao Gao, 2016), the Open Fund of State Key Laboratory of Pharmaceutical Biotechnology of Nan-jing University (KF-GN-201412), and "Challenge Cup" National Undergraduate Curricular Academic Science and Technology Works of Jinan University (16112034). We are grateful to the help of Dr. Yuan Jing-Quan at Guangxi Medicinal Botanical Garden for the isolation of TCM endophytic fungi. Assistance with the proper usage of scientific English was provided by Dr. L.J. Sparvero of the University of Pittsburgh.
Author information
Authors and Affiliations
Contributions
Prof. Dr. Hao Gao and Xin-Sheng Yao initiated the project. Prof. Dr. Hao Gao designed and coordinated the project. Mr Heng-Gang Yang, Dr. Huan Zhao, and Miss Shao-Meng Chen performed the extraction, isolation, and structural identification of the compounds. Dr. Guo-Dong Chen and Mr Jian Zou performed the quantum chemical calculation. Dr. Sheng-Ying Qin performed the paper antimicrobial assay. Dr. Chuan-Xi Wang and Miss Jiao-Jiao Li performed the isolation of fungus from A. tatarinowii. Dr. Dan Hu conducted the sequence analysis of the fungal strain (No. J13-2-12Y). Dr. Guo-Dong Chen and Miss Lang-Ming Mou performed the fermentation of the fungal strain (No. J13-2-12Y). Prof. Dr. Hao Gao and Dr. Sheng-Ying Qin wrote this paper. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, HG., Zhao, H., Li, JJ. et al. Phyllomeroterpenoids A-C, Multi-biosynthetic Pathway Derived Meroterpenoids from the TCM Endophytic Fungus Phyllosticta sp. and their Antimicrobial Activities. Sci Rep 7, 12925 (2017). https://doi.org/10.1038/s41598-017-13407-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13407-y
This article is cited by
-
Overview of fungal terpene synthases and their regulation
World Journal of Microbiology and Biotechnology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
