
Abstract
The tussock moth genus Leptocneria Butler, 1886 (Lepidoptera: Erebidae: Lymantriinae) has been considered an entirely Australian taxon that includes two species: L. reducta (Walker, 1855) and L. binotata Butler, 1886. However, we discovered a divergent lineage of Leptocneria inhabiting Flores Island, Lesser Sundas, Indonesia. Here, we describe this lineage as the third species of the genus, L. vinarskii Bolotov, Kondakov et Spitsyn sp. nov. The new species is sister to L. reducta but differs from it by dark gray marking patterns of the forewing that lack orange or dark yellow marks. The mean COI genetic distance between L. vinarskii sp. nov. and L. reducta sensu lato is 2.9%. Our findings confirm that the Wallacean region was a faunal exchange area between Sundaland and Sahul during the Pleistocene but highlight that the vicariance events may have played a crucial role in origin of the endemic faunas on the islands of East Nusa Tenggara. Additionally, we show that both Australian species most likely represent cryptic species complexes, which are in need of further taxonomic revision.
Similar content being viewed by others


Introduction
The genus Leptocneria Butler, 1886 includes two described species with an exclusively Australian distribution range1. The white cedar moth Leptocneria reducta (Walker, 1855) is famous because it is an abundant pest species, the larvae of which may cause urticarial dermatitis in humans1,2,3,4 and possibly abortions in mares5. The larvae of this species frequently defoliate white cedar trees, Melia azedarach 6,7. Additionally, these large hairy caterpillars are an important component of the diet of the Oriental Cuckoo, Cuculus saturatus Blyth, 18438. In contrast, the biological features of the other species, L. binotata Butler, 1886, have not been well studied9,10.
Although Leptocneria taxa were unknown outside Australia, in the collection of the Northern Arctic Federal University (NARFU, Arkhangelsk, Russia) we discovered a sample of moth specimens from Flores Island, Lesser Sundas, Indonesia. They are related to L. reducta, but clearly differ from it in their marking patterns. At the first glance, we assumed that they might be recent invaders from the continent, a morphological form of L. reducta. However, the DNA barcoding indicates that they actually belong to a divergent mtDNA lineage that is sister to L. reducta. Based on these findings, we concluded that this lineage is a distinct Wallacean species, and it is described herein. We also show that the Australian species most likely represent two complexes of cryptic species-level taxa, but their in-depth revision is beyond the scope of the present study.
Results
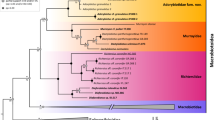
The three sequenced specimens from Flores Island share a single haplotype of the cytochrome c oxidase subunit I (COI) gene (Supplementary Table 1). The mean p-distances between this haplotype and other taxa in the genus Leptocneria are illustrated in Table 1. The Bayesian phylogenetic analysis reveals that the haplotype from Flores is sister to a large clade that contains the L. reducta sensu lato haplotypes (Fig. 1 and Supplementary Fig. 1). The Bayesian species delimitation analysis suggests that the two species from Australia, i.e., L. reducta and L. binotata, both represent species complexes, each of which comprises three molecular operational taxonomic units (MOTUs), and the haplotype from Flores belongs to a separate MOTU (Fig. 1). The mean p-distances between the MOTUs within each clade vary from 2.7 to 5.9% (Table 1). Most MOTUs share distinct distribution records that correspond to the allopatric speciation model (Fig. 2).

Biogeography and divergence times of the genus Leptocneria Butler, 1886 inferred from statistical analyses. The ultrametric chronogram was calculated under a lognormal relaxed clock model and a Yule speciation model implemented in BEAST 2.4.6 and was obtained for the COI dataset with 12 in-group haplotypes (see Supplementary Table 1 for details). Pie chaps near nodes indicate the probabilities of certain ancestral areas with respect to combined results under two different modeling approaches (S-DIVA and S-DEC). Black numbers near nodes are the mean age values, and bars are 95% confidence intervals of the estimated divergence time between lineages (Ma). A haplotype of Lymantria antennata was used as an out-group. Blue numbers near branches are Bayesian posterior probabilities inferred from MrBayes/BEAST (an asterisk indicates BPP ≥ 0.95). Solid red numbers near nodes are probabilities of species-level MOTUs (red squares) based on the highest Bayesian supported solution of the PTP species delimitation model.

Records of mtDNA lineages of Leptocneria spp. based on the DNA barcoding data (see Supplementary Table 1): L. vinarskii Bolotov, Kondakov et Spitsyn sp. nov. (1), L. reducta MOTU1 (2), L. reducta MOTU3 (3), L. reducta MOTU2 (4), L. binotata MOTU1 (5), L. binotata MOTU3 (6), and L. binotata MOTU2 (7). The map was created using ESRI ArcGIS 10 software (www.esri.com/arcgis); the topographic base of the map was created with Natural Earth Free Vector and Raster Map Data (www.naturalearthdata.com) and General Bathymetric Chart of the Oceans (www.gebco.net). Map: Mikhail Yu. Gofarov.
With respect to our combined biogeographic model and time-calibrated COI phylogeny (Fig. 1), the MRCA of L. vinarskii and Australian taxa of L. reducta complex appears to be continuously ranged in East Nusa Tenggara and Australia in the mid-Pleistocene with subsequent separation via a vicariance event (probability 96.1%; mean age 1.3 Ma, 95% HPD 0.9–1.7 Ma). Each separate biogeographic model (S-DIVA and S-DEC) also support the vicariance scenario (Supplementary Fig. 2). The origin of the crown group of the genus was placed near the Miocene – Pliocene boundary (mean age 5.1 Ma, 95% HPD 4.0–6.5 Ma). The combined scenario suggests that the Leptocneria MRCA originated somewhere in Australia or in Australia + Wallacea (probability 57.9% for Australia and 42.1% for Australia + Wallacea). The S-DEC model supports the same scenario (probability 56.8% for Australia and 43.2% for Australia + Wallacea), whereas the S-DIVA model indicates the possible primary role of Australia (probability 72.5%) (Supplementary Fig. 2).
Taxonomy
Family Erebidae Leach, [1815]
Subfamily Lymantriinae Hampson, 1893
Genus Leptocneria Butler, 1886
Type Species: Leptocneria binotata Butler, 1886
Leptocneria vinarskii Bolotov, Kondakov et Spitsyn sp. nov.
Type material. Holotype male, INDONESIA, Lesser Sundas, East Nusa Tenggara, Flores Island: Labuan Bajo, Komodo Ecolodge, 8°31’21”S, 119°52’16”E, garden and grasslands on the sea coast, 13–20.i.2015, local coll. leg. (NARFU, voucher no. Sph0589). Paratypes: 3♀, INDONESIA, Lesser Sundas, East Nusa Tenggara, Flores Island: Labuan Bajo, Komodo Ecolodge, garden and grasslands on the sea coast, 8°31’21”S, 119°52’16”E, 13–24.i.2015, local coll. leg. (NARFU, voucher nos. Sph588, Sph700, and Sph701) (Fig. 3).

Leptocneria vinarskii Bolotov, Kondakov et Spitsyn sp. nov. Holotype male (specimen no. Sph0589, reference COI sequence no. MF036688): (a) upper side, and (b) underside. Paratype female (specimen no. Sph588, reference COI sequence no. MF036689): (c) upper side, and (d) underside (scale bar = 5 mm). Male genitalia (holotype): (e) genitalia, and (f) aedeagus (scale bar = 1 mm). (g) Type locality: Labuan Bajo, Komodo Ecolodge, garden and grasslands on the sea coast. Photos: Vitaly M. Spitsyn (a–f) and Yulia S. Kolosova (g).
DNA barcoding: Reference sequences in GenBank: MF036687, MF036688, and MF036689. The mean COI p-distance between the new species and L. reducta sensu lato is 2.9 ± 0.6%, and that between the new species and L. binotata sensu lato is 9.6 ± 1.3% (Table 1).
Etymology. This new species is dedicated to Dr. Maxim V. Vinarski, a well-known Russian zoologist.
Diagnosis. The new species is similar to L. reducta sensu lato but differs from it by dark gray marking patterns of the forewing, which lack orange or dark yellow marks.
Description. Head: Male and female antennae bipectinate. Eyes black, without hairs. Frons gray. Labial palpi longer than eye diameter, dark gray. Thorax: Thorax and legs uniformly dark gray. Forewing length: male 18 mm; female 23–26 mm. Upper side of male forewing gray, with dark gray markings: unclear marks between veins in marginal area, diffused zigzag postdiscal line, large rounded discal spot, broadly dark along costal area. Upper side of male hindwing light gray, with unclear rounded gray discal spot, darkness in apical area and unclear grey marks between veins in marginal area. Underside of male forewing gray, with dark gray marks between veins in marginal area, broadly dark in postdiscal and costal areas, with large diffuse discal spot. Underside of male hindwing light gray, with vague rounded gray discal spot, and small gray spots between veins in marginal area. Upper side of female forewing gray, with diffuse dark gray markings: vague marks between veins in marginal area, large discal and postbasal spots, and narrowly dark along costal and dorsal margins. Upper side of female hindwing gray, with small, diffuse discal spot. Undersides of female wings uniformly gray, slightly dark in cell of the forewing. Fringes of both wings gray. Abdomen: Uniformly dark gray. Male genitalia: Uncus long, straight, tapering posteriorly to pointed tip. Valva short, with broad basal half and narrow, acuminate distal half. Saccus conical. Aedeagus long, strongly concave, expanded at base. Coremata absent. Female genitalia: not examined.
Distribution. West Flores; known only from the type locality, but may also inhabit other East Nusa Tenggara islands (e.g., Timor, Sumba, and Sumbawa), whose lepidopteran faunas are poorly known.
Habitat. Lowland semi-natural habitats near the sea coast.
Discussion
Our results reveal that the number of species in the genus Leptocneria was largely underestimated, because each of the previously described species in the genus most likely represents a species complex (Fig. 1 and Supplementary Fig. 1). Although a revision of the cryptic taxa from Australia is well beyond the scope of this study, we could suggest that at least part of the lineages most likely arose via allopatric speciation driven by some inland barriers, e.g., continuous desert areas (Fig. 2). However, we discovered that the distributional range of this genus, which was considered endemic to Australia1, extends into the islands of East Nusa Tenggara. The distant lineage from Flores Island belongs to L. vinarskii sp. nov., a species that is new to science (Fig. 3). This lineage shares a common ancestor with L. reducta sensu lato, which most likely spread to East Nusa Tenggara from Australia through the drying Sahul Shelf (Fig. 1). In accordance with the results of our biogeographic modeling and time-calibrated phylogenetic analyses, we could suggest that these taxa were likely separated via a vicariance event in the mid-Pleistocene, approximately 1.3 Ma ago. The ancestral area of the Leptocneria MRCA remains uncertain, because different models placed it in Australia or in Australia + Wallacea, although the primary role of the Sahul region for subsequent diversification of the genus is not in doubt.
The lepidopteran fauna of the Lesser Sunda Islands is poorly known. A few available sources reveal that the faunas of these islands are largely of Asian origin11,12,13,14. Examples of moth taxa with clear Asian affinities inhabiting the East Nusa Tenggara Islands are common among the Erebidae, Lycaenidae, Papilionidae, and other families11,15,16,17,18,19. In contrast, the spread of Australian moth taxa into Wallacea is less well-known, although examples have been recorded among the Sphingidae20 and Lasiocampidae21,22. The biogeographic analysis of birdwing butterflies (Papilionidae) suggests that Wallacea was the source of numerous dispersal events towards neighboring areas (Sahul and Sunda)11. Our novel discovery confirms that the Wallacean region was a faunal exchange area between Sundaland and Sahul during the Pleistocene14, but highlights that the vicariance events may have played a crucial role in origin of the endemic faunas on the islands of East Nusa Tenggara.
Methods
Taxon sampling and laboratory protocols
The sequence data set that combine our materials and published data includes a total of 48 sequences of Leptocneria spp. (Supplementary Table 1). Available COI sequences were obtained from the Barcoding of Life Identification System (BOLD IDS) database and from NCBI’s GenBank23,24. The majority of these specimens was sequenced under a comprehensive analysis of Lepidoptera from the Australian National Insect Collection25. For molecular analyses, we used three specimens of L. vinarskii Bolotov, Kondakov et Spitsyn sp. nov. from the collection of the Northern Arctic Federal University (NARFU), Arkhangelsk, Russia. The total DNA was extracted from a single leg of each dry specimen using a standard phenol/chloroform procedure26. The standard primers LepF and LepR were used for the amplification of 660-bp-long barcode fragments of the COI gene27. The PCR mix contained approximately 200 ng of total cellular DNA, 10 pmol of each primer, 200 μmol of each dNTP, 2.5 μl of PCR buffer (with 10 × 2 mmol MgCl2), 0.8 units Taq DNA polymerase (SibEnzyme Ltd., Novosibirsk, Russia), and H2O added for a final volume of 25 μl. Thermocycling included one cycle at 95 °C (4 min), followed by 38–40 cycles of 95 °C (50 sec), 50 °C (50 sec), and 72 °C (50 sec) and a final extension at 72 °C (5 min). Forward and reverse sequencing was performed on an automatic sequencer (ABI PRISM® 3730, Applied Biosystems) using an ABI PRISM® BigDye™ Terminator v. 3.1 reagent kit. Resulting sequences were checked manually using a sequence alignment editor (BioEdit version 7.2.5)28.
Sequence alignment and phylogenetic analysis
The alignment of sequences was performed using the ClustalW algorithm of MEGA629. For phylogenetic analyses, the sequence data set was collapsed into 12 unique COI haplotypes of Leptocneria spp. (657 bp in length) using an online FASTA sequence toolbox, FaBox v. 1.4130, with subsequent checking via a p-distance matrix of MEGA6 (we used uncorrected pairwise genetic distances)29. As an out-group, a haplotype of Lymantria antennata Walker, 1855 was used (Supplementary Table 1). The lacking sites were treated as missing data. The best models of sequence evolution as suggested by the corrected Akaike Information Criterion of MEGA629 were as follows: 1st codon of COI: TN93+G (G = 0.31), 2nd codon of COI: TN93, and 3rd codon of COI: HKY+I (I = 0.12). Phylogenetic relationships were reconstructed based on Bayesian inference implemented in MrBayes v. 3.2.631. The analyses were performed using the following parameters: nchains = 4, nruns = 2, samplefreq = 1000, temp = 0.1; 10% of the sampled trees were discarded as burn-in (pre-convergence part). Runs were conducted for 3 million generations. Convergence of the MCMC chains to the stationary distribution was checked visually based on the plotted posterior estimates using a MCMC trace analysis tool (Tracer v1.6)32. Calculations were performed at the San Diego Supercomputer Center through the CIPRES Science Gateway33.
Species delimitation
To delimit prospective species-level units, we used a molecular approach based on the concept of MOTUs34,35. The MOTUs were separated using the Poisson Tree Processes (PTP) model to infer putative species boundaries on a phylogenetic input tree inferred from a Bayesian analysis of the COI haplotype sequences36. We used a Bayesian implementation of the PTP model for species delimitation through an online bPTP server (http://species.h-its.org/ptp) with 100,000 MCMC generations and 10% burn-in36. The out-group haplotype was removed from the input tree using an appropriate option of the server.
Divergence time estimates
We estimated the acceptance of a molecular clock approach to our multi-gene data set using the Tajima’s relative rate test of MEGA629, which is not reject the null hypothesis of equal rates between lineages (P > 0.05 in all possible combinations). Hypothetical divergence times were estimated in BEAST 2 v. 2.4.6 using a lognormal relaxed clock algorithm with the Yule speciation model as the tree prior37. Calculations were performed at the San Diego Supercomputer Center through the CIPRES Science Gateway33. We specified similar settings to three partitions (3 codons of COI) as in the MrBayes analyses (see above). To dating the phylogeny, a substitution rate of 1.78% per million years for COI was applied, which is the most reliable estimation of the mean evolutionary rate in insects38. Four replicate searches were conducted, each with 25 million generations. The trees were sampled every 1,000th generation. The log files were checked visually with Tracer v. 1.6 for an assessment of the convergence of the MCMC chains and the effective sample size (ESS) of parameters32. All ESS values were recorded as >400; the posterior distributions were similar to the prior distributions. The resulting tree files from four independent analyses were compiled with LogCombiner v. 2.1.337. The first 10% of trees were discarded as an appropriate burn-in. The maximum clade credibility tree was obtained by using TreeAnnotator v. 2.1.337.
Ancestral area reconstructions
We tested ancestral area patterns using two different approaches, i.e., Statistical Dispersal-Vicariance Analysis (S-DIVA) and Statistical Dispersal-Extinction Cladogenesis (S-DEC) implemented in RASP v. 3.239. For the ancestral area reconstruction, we used the set of 90,004 post-burn-in binary trees that were combined from four runs of BEAST v. 2.4.6 (see above). As a condensed tree, we used the user-specified consensus tree, which was obtained based on this set of trees using TreeAnnotator v. 2.1.3 (see above). From both of the tree data sets, out-group sequence was removed using the appropriate option of RASP v. 3.2. We coded two possible distribution areas of the in-group taxa as follows: (a) Australia (Sahul), and (b) Wallacea. The S-DIVA models were calculated with the following parameters: max areas = 2; allow reconstruction with max reconstructions = 100; max reconstructions for final tree = 1000; and allowing extinctions. The S-DEC analyses were run with default settings and max areas = 2. In addition to the evaluations obtained from each analysis separately, we used generalized results of the two modeling approaches, which were combined using an algorithm implemented in RASP v. 3.2.
Nomenclatural acts
The electronic edition of this article conforms to the requirements of the amended International Code of Zoological Nomenclature, and hence the new name contained herein is available under that Code from the electronic edition of this article. This published work and the nomenclatural acts it contains have been registered in ZooBank (http://zoobank.org/), the online registration system for the ICZN. The LSID for this publication is: urn:lsid:zoobank.org:pub:435C007C-6B18-4953-9BE5-5823F502D613. The electronic edition of this paper was published in a journal with an ISSN, and has been archived and is available from PubMed Central.
Data availability
The COI sequences generated during this study are available from GenBank. Accession number for each specimen is presented in Supplementary Table 1. The type specimens of the new species are available in the collection of the Northern Arctic Federal University (NARFU), Arkhangelsk, Russia (voucher nos. Sph0589 [holotype], Sph588, Sph700, and Sph701 [paratypes]).
References
Common, I. F. B. Moths of Australia. Melbourne: Melbourne University Publishing (1990).
Hossler, E. W. Caterpillars and moths: Part II. Dermatologic manifestations of encounters with Lepidoptera. Journal of the American Academy of Dermatology 62, 13–28, https://doi.org/10.1016/j.jaad.2009.08.060 (2010).
Nikitin, M. I. On the seasonal distribution of some common moths (Lep., Heterocera) at Cabramatta, NSW. Journal of the Entomological Society of Australia (NSW) 2, 37–39 (1965).
Villas-Boasa, I. M., Alvarez-Floresb, M. P., Chudzinski-Tavassib, A. M. & Tambourgia, D. V. Envenomation by caterpillars. Clinical Toxinology; https://doi.org/10.1007/978-94-007-6288-6_57-1 (2016)
Todhunter, K. H. et al. Equine amnionitis and fetal loss: The case definition for an unrecognised cause of abortion in mares. Australian Veterinary Journal 87, 35–38, https://doi.org/10.1111/j.1751-0813.2008.00386.x (2009).
King, J. & Lawson, S. 8. Insect pests and diseases of rainforest timber species grown in plantations. In: Erskine, P.D., Lamb, D. & Bristow, M. (Eds.) Reforestation in the Tropics and Subtropics of Australia Using Rainforest Tree Species. Rural Industries Research and Development Corporation, Canberra, pp. 114–128 (2005).
Richardson, C. A. Notes on some insects intercepted entering New Zealand in 1975. New Zealand Entomologist 6, 309–311, https://doi.org/10.1080/00779962.1977.9722273 (1977).
Bravery, J. A. Field notes on the Oriental Cuckoo. Cuculus saturatus. Emu - Austral Ornithology 66, 267–271 (1967).
Butler, A. G. X. V. Descriptions of 21 new genera and 103 new species of Lepidoptera - Heterocera from the Australian Region. Transactions of the Royal Entomological Society of London 34, 381–442 (1886).
Turner, A. J. Revision of Australian Lepidoptera—Liparidae. Proceedings of the Linnean Society of New South Wales 45, 474–499 (1920).
Condamine, F. L. et al. Deciphering the evolution of birdwing butterflies 150 years after Alfred Russel Wallace. Scientific Reports 5, 11860, https://doi.org/10.1038/srep11860 (2015).
Holloway, J. D. Taxonomic Appendix. In: Barlow, H.S. An introduction to the moths of South East Asia. Kuala Lumpur: The Author, pp. 174–271 (1982).
Holloway, J. D. The Moths of Borneo: family Arctiidae, subfamily Lithosiinae. Malaysian Nature Journal 55, 279–458 (2001).
Lohman, D. J. et al. Biogeography of the Indo-Australian archipelago. Annual Review of Ecology, Evolution, and Systematics 42, 205–226, https://doi.org/10.1146/annurev-ecolsys-102710-145001 (2011).
Černý, K. A contribution to the knowledge of the genus Cyana Walker in the South East Asia (Lepidoptera: Erebidae, Arctiinae, Lithosiinae). Entomofauna 37, 421–436 (2016).
Spitsyn, V. M., Bolotov, I. N., Gofarov, M. Y. & Bolotov, N. I. First record of the genus Aethalida Walker, 1865 (Lepidoptera: Erebidae: Arctiinae) from Flores Island, East Nusa Tenggara, Indonesia. Ecologica Montenegrina 6, 56–60 (2016).
Bito, D. An alien in an archipelago: Spathodea campanulata and the geographic variability of its moth (Lepidoptera) communities in the New Guinea and Bismarck Islands. Journal of Biogeography 34, 769–778, https://doi.org/10.1111/j.1365-2699.2006.01652.x (2007).
Dubatolov, V. V. Tiger-moths of Eurasia (Lepidoptera, Arctiidae) (Nyctemerini by R. de Vos & V. V. Dubatolov). Neue Entomologische Nachrichten 65, 1–106 (2010).
Lohman, D. J., Peggie, D., Pierce, N. E. & Meier, R. Phylogeography and genetic diversity of a widespread Old World butterfly, Lampides boeticus (Lepidoptera: Lycaenidae). BMC Evolutionary Biology 8, 301, https://doi.org/10.1186/1471-2148-8-301 (2008).
Rougerie, R. et al. Australian Sphingidae - DNA barcodes challenge current species boundaries and distributions. PLoS One 9, e101108, https://doi.org/10.1371/journal.pone.0101108 (2014).
Zolotuhin, V. V. Peculiarities of island endemism in Lasiocampidae (Lepidoptera). Entomological Review 89, 34–45, https://doi.org/10.1134/S0013873809010060 (2009).
Zolotuhin, V. V. & Witt, T. J. Contribution to the knowledge of Indonesian Lasiocampidae (Lepidoptera). Tinea 19, 59–68 (2005).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of Molecular Biology 215, 403–410, https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Ratnasingham, S. & Hebert, P. D. N. A DNA-Based Registry for All Animal Species: The Barcode Index Number (BIN) System. PLoS ONE 8, e66213, https://doi.org/10.1371/journal.pone.0066213 (2013).
Hebert, P. D. et al. A DNA ‘Barcode Blitz’: Rapid digitization and sequencing of a natural history collection. PLoS ONE 8, e68535, https://doi.org/10.1371/journal.pone.0068535 (2013).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular Cloning: A Laboratory Manual (2nd ed.). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp. 10.51–10.67 (1989).
Hajibabaei, M., Janzen, D. H., Burns, J. M., Hallwachs, W. & Hebert, P. D. N. DNA barcodes distinguish species of tropical Lepidoptera. Proceedings of the National Academy of Sciences of the United States of America 103, 968–971, https://doi.org/10.1073/pnas.0510466103 (2006).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41, 95–98 (1999).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular Biology and Evolution 30, 2725–2729, https://doi.org/10.1093/molbev/mst197 (2013).
Villesen, P. FaBox: an online toolbox for FASTA sequences. Molecular Ecology Notes 7, 965–968, https://doi.org/10.1111/j.1471-8286.2007.01821.x (2007).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Systematic Biology 61, 539–542, https://doi.org/10.1093/sysbio/sys029 (2012).
Rambaut, A., Suchard, M.A., Xie, D. & Drummond, A.J. Tracer v1.6 http://beast.bio.ed.ac.uk/Tracer (2014).
Miller, M., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Gateway ComputingEnvironments Workshop (GCE), New Orleans Convention Center New Orleans, LA, USA, pp. 1–8 (2010).
Blaxter, M. et al. Defining operational taxonomic units using DNA barcode data. Philosophical Transactions of the Royal Society B: Biological Sciences 360, 1935–1943, https://doi.org/10.1098/rstb.2005.1725 (2005).
De Queiroz, K. Species concepts and species delimitation. Systematic Biology 56, 879–886, https://doi.org/10.1080/10635150701701083 (2007).
Zhang, J., Kapli, P., Pavlidis, P. & Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29, 2869–2876, https://doi.org/10.1093/bioinformatics/btt499 (2013).
Bouckaert, R. et al. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Computational Biology 10, e1003537, https://doi.org/10.1371/journal.pcbi.1003537 (2014).
Papadopoulou, A., Anastasiou, I. & Vogler, A. P. Revisiting the insect mitochondrial molecular clock: the mid-Aegean trench calibration. Molecular Biology and Evolution 27, 1659–1672, https://doi.org/10.1093/molbev/msq051 (2010).
Yu, Y., Harris, A. J., Blair, C. & He, X. J. RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Molecular Phylogenetics and Evolution 87, 46–49, https://doi.org/10.1016/j.ympev.2015.03.008 (2015).
Acknowledgements
We thank the Associate Editor Dr. Mark de Bruyn, Dr. Camiel Doorenweerd, and two anonymous reviewers for their helpful comments. This work was partly funded by grants from the President of Russia Grant Council (project no. MD-7660.2016.5), Russian Ministry of Education and Science (project no. 6.2343.2017/4.6), Federal Agency for Scientific Organizations (project no. 0410-2014-0028), and Northern Arctic Federal University. We are grateful to Mr. Mikel Albarran Valle (Labuan Bajo, Indonesia) for his great help during this study.
Author information
Authors and Affiliations
Contributions
I.N.B. developed the concept of the study. A.V.K. designed and carried out molecular analyses. M.Y.G. created the map. V.M.S. pictured the specimens. Y.S.K. photographed the habitat. I.N.B. wrote the paper, with input from A.V.K., V.M.S., M.Y.G. and Y.S.K. All authors discussed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bolotov, I.N., Kondakov, A.V., Spitsyn, V.M. et al. Leptocneria vinarskii sp. nov. (Lepidoptera: Erebidae: Lymantriinae), an overlooked Wallacean lineage of the Australian genus. Sci Rep 7, 12430 (2017). https://doi.org/10.1038/s41598-017-12797-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12797-3
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
