
Abstract
Astragaloside II (AS II) extracted from Astragalus membranaceus has been reported to promote tissue wound repair. However, the effect of AS II on inflammatory bowel disease is unknown. We investigated the effects and mechanism of AS II on intestinal wound healing in both in vitro and in vivo models. Human intestinal Caco-2 cells were treated with multiple concentrations of AS II to assess cell proliferation, scratch wound closure, L-arginine uptake, cationic amino acid transporter activity, and activation of the mTOR signaling pathway. These effects were also measured in a mouse model of colitis. AS II promoted wound closure and increased cell proliferation, L-arginine uptake, CAT1 and CAT2 protein levels, total protein synthesis, and phosphorylation of mTOR, S6K, and 4E-BP1 in Caco-2 cells. These effects were suppressed by lysine or rapamycin treatment, suggesting that the enhanced arginine uptake mediates AS II-induced wound healing. Similar results were also observed in vivo. Our findings indicate that AS II can contribute to epithelial barrier repair following intestinal injury, and may offer a therapeutic avenue in treating irritable bowel disease.
Similar content being viewed by others


Introduction
Inflammatory bowel disease (IBD) is a chronic gastrointestinal disorder, which can manifest as ulcerative colitis and Crohn’s disease1. Clinical symptoms include weight loss, abdominal pain, diarrhea, and bleeding2, and continuous mucosal inflammation can lead to intestinal fibrosis and may subsequently progress and develop into colon cancer3. The prevalence of IBD has been reported as 200 per 100,000 in the US, and this number is increasing; IBD has now become a global health issue as more countries are adopting a Western diet4. Although the precise mechanisms of IBD are still unknown, most studies concur that IBD is associated with hereditary, infectious, environmental, and auto-immune factors. The integrity of the intestinal epithelial barrier plays a role in IBD progression5. Recent studies have indicated that restoration of the epithelial barrier integrity is an important healing response in IBD and other intestinal disorders6,7,8,9,10. Thus, the repair of the intestinal epithelial barrier may be a promising therapeutic strategy in IBD. Current medications, such as non-steroidal anti-inflammatory drugs, steroids, and immunodulators, are limited in their application because of poor efficacy and adverse effects10. Therefore, a new effective therapy for IBD is needed.
Recovery of the epithelial barrier is crucial in the treatment of colitis. L-arginine (L-Arg) is involved in protein synthesis and regulation of many essential cellular functions, including immune response, hormone secretion, and wound healing11. In addition, L-Arg and its metabolite ornithine promote colonic epithelial wound repair by enhancing cell proliferation and collagen deposition12. L-Arg uptake has been shown to occur primarily by cationic amino acid transporter 2 (CAT 2), and is an important process in the restoration of colonic epithelial cells10. This is also confirmed by evidence that L-Arg supplementation suppressed intestinal permeability and improved IBD symptoms by enhancing wound healing in an IBD rodent model7. Protein metabolism in intestinal mucosa is essential for gut homeostasis and maintenance of the epithelial barrier13. L-Arg increases intestinal protein synthesis and epithelial repair by activating the mechanistic target of rapamycin (mTOR) signaling pathway14. Once activated, mTOR phosphorylates its downstream targets, ribosomal protein S6 kinase (p70 S6K) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), thereby promoting mRNA translation, protein synthesis, and cell growth15. In contrast, blockage of the mTOR pathway suppresses intestinal cell migration16. In this way, L-Arg contributes to wound healing and protein synthesis, while significantly enhancing mTOR signaling; this pathway may be a promising agent in intestinal wound closure.
Radix Astragali is a well-known medicinal herb for reinforcing Qi (the vital energy) in traditional Chinese medicine, which considers it to possess immunomodulatory, wound-healing, anti-inflammatory, anti-aging, anti-oxidant, and hypoglycemic properties17. Astragalus membranaceus contains a variety of compounds, including polysaccharides, flavonoids, and saponins. Astragaloside II (AS II; Fig. 1A) is one of the major cycloartane-type triterpene glycosides extracted from Radix Astragali18, and has recently been reported to be a potential adjunctive agent in cancer chemotherapy19, enhancement of osteogenesis20, and modulation of T cell activation21. However, the effects and underlying mechanism of AS II on intestinal wound healing are unknown. In the present study, we examined the effect of AS II in repair and restoration of intestinal epithelial barrier function and the signaling mechanism involved, both in vitro and in vivo.

Effects of astragaloside II (AS II) on cell proliferation and scratch wound closure in Caco-2 cells. (A) Chemical structure of AS II. (B) Caco-2 cells were treated with 0.01, 0.1, and 1 μM AS II for 24 hr. Cell proliferation was measured using a CCK-8 kit. (C) The Caco-2 cell monolayers were scratched and incubated with the indicated concentrations of AS II. Wound closure was photographed and quantified over time. Data represent mean ± SEM (n = 6). *p < 0.05, **p < 0.01, and ***p < 0.001 versus the untreated control.
Results
AS II promotes cell proliferation and scratch wound closure in Caco-2 cells
We first investigated the effect of AS II on cell proliferation. AS II increased cell proliferation (fold-change compared with control: 1.11 ± 0.03 for 0.01 μM, p < 0.05; 1.22 ± 0.03 for 0.1 μM, p < 0.001; 1.16 ± 0.03 for 1 μM, p < 0.001) (Fig. 1B). AS II improved scratch wound closure in Caco-2 cells in a time-dependent manner (Fig. 1C); the maximum effect of scratch wound closure was observed at 0.1 μM. Forty-eight hours after initiating the scratch wound assay, scratch wound percent closure increased from 30.77 ± 2.13 to 41.42 ± 2.09 (0.01 μM, p < 0.01), 42.17 ± 1.42 (0.1 μM, p < 0.001), and 39.79 ± 1.61 (1 μM, p < 0.01) (Fig. 1C).
AS II increases L-Arg uptake and CAT protein levels in Caco-2 cells
To assess the effects of AS II on L-Arg uptake, we treated Caco-2 cells with AS II. As shown in Fig. 2A, 0.1 μM of AS II significantly increased L-Arg cellular uptake between 0.5 and 24 hr compared with the control, reaching 104.73 ± 3.90 pmol/mg protein/min 6 hr after treatment (p < 0.001). The stimulatory effect of AS II was observed in the concentrations tested; the greatest effect was seen in the 0.1-μM treatment (108.18 ± 8.02 pmol/mg protein/min, p < 0.001 compared with the control; Fig. 2B). To identify the L-Arg transporters involved, we measured the expression of CAT1 and CAT2, and found that 0.1 μM AS II significantly increased both CAT1 and CAT2 expression (Fig. 2C and D) between 6 and 48 hr after treatment. A higher concentration of AS II (1 μM) also increased CAT1 and CAT2 protein levels (1.45 ± 0.18 fold-increase over control for CAT1, p < 0.05; 1.55 ± 0.05 fold-increase over control for CAT2, p < 0.05; Fig. 2E and F).

Effects of astragaloside II (AS II) on L-arginine (L-Arg) uptake and cationic amino acid transporter (CAT) expression. Differentiated Caco-2 cells were incubated with AS II and then analyzed for L-Arg accumulation at the indicated time periods (A) and concentrations (B). CAT1 and CAT2 protein levels in Caco-2 cells were assessed by western blotting at different time points (C and D) and concentrations (E and F) of AS II treatment. Data represent mean ± SEM (n = 6 for L-Arg uptake, n = 3 for western blotting). *p < 0.05, **p < 0.01, and ***p < 0.001 versus the untreated control.
AS II activates the mTOR pathway and enhances protein synthesis in Caco-2 cells
We studied the effect of AS II on mTOR activation and protein synthesis. AS II (0.1 μM) increased phosphorylated mTOR levels, especially 1 hr after treatment (1.44 ± 0.02 fold-increase over control, p < 0.01; Fig. 3A and B). AS II also promoted the phosphorylation of mTOR’s downstream targets S6K and 4EBP1, especially 3 and 6 hr after treatment (p < 0.01 for p-S6K and p < 0.05 for p-4EBP1 compared with the control; Fig. 3A,C and D). We then investigated the effect of AS II on cellular protein synthesis. AS II significantly increased protein synthesis in both a time- and concentration-dependent manner (Fig. 3E and F). These findings indicate AS II increases both the mTOR signal pathway and protein synthesis in Caco-2 cells.

Effects of astragaloside II (AS II) on the mTOR signaling pathway and on protein synthesis. Caco-2 cells were incubated with 0.1 μM AS II and then harvested for western blotting (A). Levels of p-mTOR/mTOR (B), p-S6K/S6K (C), and p-4E-BP1/4E-BP1 (D) were quantified. Protein synthesis was assayed by [3H]-leucine incorporation and quantified by time (E) and concentration (at 24 hr, F). Data represent mean ± SEM (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001 versus the untreated control.
Lysine and rapamycin both suppress the effects of AS II on the mTOR signaling pathway
To investigate the roles of L-Arg uptake and mTOR signaling in the wound-healing activity of AS II, we used lysine (a competitive inhibitor of CAT1 and CAT2 for L-Arg uptake) and rapamycin (the mTORC1 inhibitor). Pretreatment with lysine reduced AS II-upregulated scratch wound closure (from 40.58 ± 1.48 to 32.10 ± 1.54%; p < 0.01; Fig. 4A), protein synthesis (from 120.60 ± 2.48 to 94.88 ± 2.09%; p < 0.001; Fig. 4B), p-mTOR (from 1.18 to 0.93 fold-increase over control; Fig. 4C), and p-S6K (from 1.43 to 0.97 fold-increase over control; Fig. 4D). Rapamycin exerted similar effects on AS II-upregulated scratch wound closure (from 29.36 ± 1.01 to 18.43 ± 0.52%; p < 0.05; Fig. 4E), protein synthesis (from 118.14 ± 1.49 to 92.20 ± 2.56% compared with control; p < 0.001; Fig. 4F), and p-S6K (from 1.71 ± 0.08 to 0.21 ± 0.17 fold-increase over control; p < 0.01; Fig. 4G). The results indicated that both upregulation of the L-Arg uptake and mTOR activation are necessary for AS II-mediated wound healing.

Effects of L-lysine and rapamycin on wound closure and protein synthesis in astragaloside II (AS II)-treated Caco-2 cells. Cells were incubated with L-lysine (20 mM) or rapamycin (100 nM) 30 min prior to treatment with 0.1 μM AS II. Scratch wound closure and protein synthesis were quantified after 36 hr (A) and 24 hr (B), respectively. Levels of p-mTOR/mTOR (C) and p-S6K/S6K (D) were analyzed in the L-lysine-treated cells. Similarly, scratch wound closure (E), protein synthesis (F), and p-S6K/S6K levels (G) were analyzed in the rapamycin-treated cells. Data represent mean ± SEM (n = 3). *p < 0.05 and **p < 0.01 versus the untreated control; # p < 0.05 versus the AS II-treated cells.
AS II attenuates 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis in mice
To assess the in vivo effects of AS II, we established a TNBS-induced mouse model of colitis. We found that treatment with AS II attenuated TNBS-induced weight loss (from 85.75 ± 2.54 to 94.85 ± 1.78% compared with the control [100%], p < 0.01; Fig. 5A), and affected the length of the small intestine (from 28.46 ± 1.40 to 31.88 ± 0.85 cm, p < 0.05; Fig. 5B), length of the large intestine (from 5.40 ± 0.18 to 6.22 ± 0.22 cm, p < 0.05; Fig. 5C), and myeloperoxidase (MPO) activity in the small intestine (from 1.15 ± 0.30 to 0.35 ± 0.08 unit/mg protein, p < 0.01; Fig. 5D). MPO activity in the large intestine (Fig. 5E) and epithelial barrier permeability (Fig. 5F) were also measured, but did not differ statistically from the control. AS II significantly increased L-Arg uptake in the small intestine (from 500.60 ± 64.17 to 731.29 ± 75.82 pmol/min/mg protein, p < 0.05; Fig. 5G). Slight but non-significant increase was observed in the colon (Fig. 5H). AS II also increased CAT1 and CAT2 levels in the small intestine (Fig. 5I) and CAT1 levels in the colon (Fig. 5J). Colon histology appeared altered (Fig. 5K).

Effects of astragaloside II (AS II) on a 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced mouse model of colitis. (A) Body weight. (B) Length of the small intestine. (C) Length of the colon. Myeloperoxidase (MPO) activity was examined in both the small intestine (D) and the colon (E). (F) mucosal permeability. (G and H) L-arginine (L-Arg) uptake. (I and J) Levels of cationic amino acid transporters (CATs). (K) Histological examination was performed by photomicrography (original magnification at 40× and 100×). Data represent mean ± SEM (n = 6). *p < 0.05, ***p < 0.01, and ***p < 0.001 versus the untreated control; # p < 0.05 and ## p < 0.01 versus the AS II-treated cells.
Discussion
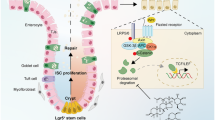
In this study, we examined the effects of AS II, one of the major bioactive components of A. membranaceus, on repair and restoration of intestinal epithelial barrier function, and investigated the signaling mechanism involved. We found that AS II can promote scratch wound closure, cell proliferation, and arginine uptake, and can induce the arginine transporters CAT1 and CAT2 in differentiated human intestinal Caco-2 cells. In addition, AS II enhanced the phosphorylation of mTOR, S6K, and 4E-BP1, while protein synthesis increased significantly. The effects of AS II on wound closure were also confirmed using L-lysine (a competitive inhibitor of L-Arg uptake) and rapamycin (a specific mTOR inhibitor), as both of these inhibitors suppressed the effect of AS II on wound closure. These results suggest that L-Arg uptake and mTOR signaling activation are involved in AS II-induced wound healing. The effect of 0.1 μM AS II was greater than that of 1 μM AS II on cell proliferation, wound closure, L-Arg uptake, CAT1 expression, and leucine incorporation. These findings indicate that 0.1 μM is an effective concentration. A 1-μM treatment appeared to exert adverse effects or to inhibit the beneficial effects. The higher concentration may induce or interfere with certain cellular activities. The dose independent activity has been reported in different natural product22. In a TNBS-induced mouse colitis model, AS II was shown to ameliorate the severity of colitis symptoms such as weight loss, reduction in intestinal length, intestinal inflammation, and increased mucosal permeability. AS II also increased intestinal L-Arg uptake and mucosal CAT protein levels. Our findings indicate that AS II promotes intestinal epithelial healing through increased L-Arg uptake and protein synthesis, which are likely mediated by the increased expression of L-Arg transporters and activation of the mTOR signaling pathway, respectively. Figure 6 displays our proposed mechanism of AS II action.

Proposed mechanism of astragaloside II (AS II) action on intestinal epithelial wound repair.
A recent strategy for managing IBD has been to improve the integrity of the intestinal epithelial barrier and prevent recurrence of intestinal inflammation, especially in Crohn’s disease23. In traditional Chinese medicine, Radix Astragali is a tonic herb used in a variety of inflammatory and immune diseases, as well as for wound healing21. In the present study, we showed that AS II can promote intestinal epithelial wound healing via the mTOR pathway in vitro and in vivo. IBD pathogenesis is a complex process, which involves the immune response and inflammation1. Crude extract of Radix Astragali has been shown to exert protective and anti-inflammatory effects in several animal models of colitis24,25,26. To our knowledge, this is the first study to report the beneficial effects of AS II, a major bioactive component of Astragalus, in a TNBS-induced animal model of colitis. Our findings provide a better understanding of the therapeutic potential of Radix Astragali in IBD. We also showed that MPO activity was inhibited in AS II-treated mice, indicating its anti-inflammatory potential. Future work should assess whether AS II can attenuate IBD symptoms by modulating the immune response.
Astragalus products have been used as immunomodulatory and wound-healing agents in traditional Chinese medicine17. However, the detailed mechanisms and bioactive components are largely unknown. Radix Astragali contains a variety of compounds such as polysaccharides, flavonoids, isoflavonoids, and polyphenols. Among these, astragalosides have been shown to promote wound-healing activity in vitro and in vivo 27. To date, most studies have focused on the pharmacological effects of AS IV, a major astragaloside, which has been shown to improve wound repair and diminish scarring in burn wounds28. However, few studies have investigated AS II’s effects on intestinal epithelial healing. The structure of AS II is similar to that of AS IV, apart from an additional acetyl group at C-2 of the xylose residues. We found that AS II improved intestinal epithelial repair by enhancing L-Arg uptake and activating the mTOR pathway. AS II has been shown to exert better T cell immune-stimulating effects than AS IV21. This suggests that AS II may be a more potent immune stimulator than AS IV. Current drugs indicated for IBD treatment include anti-inflammatory drugs, immunosuppressives, antibiotics, and biologic agents. These agents can cause side effects such as gastrointestinal symptoms, hepatotoxicity, and renal toxicity, limiting their clinical applications29. Radix Astragali is considered a top-grade herb in traditional Chinese medicine, meaning a safe and nontoxic herbal medicine30. Astragalus supplementation (90 g daily) has been reported to have low toxicity and no significant adverse effects31. AS II may therefore be a safe and promising compound for treating IBD or immune disorders.
L-Arg has been shown to promote wound healing in skin, intestinal epithelial tissues, and in vivo IBD models7,32,33. In the present study, AS II improved L-Arg uptake in both cultured Caco-2 cells and colonic tissue, and increased CAT1 and CAT2 levels. Cellular uptake of L-Arg through CAT2 is important for colonic epithelial wound repair10. One study indicated that 12-O-tetradecanoylphorbol-13-acetate increased L-Arg uptake and CAT levels by activating the protein kinase C (PKC) signaling pathway in intestinal epithelial cells34. Whether AS II promotes CAT expression via the PKC signaling pathway requires further study. High doses of L-Arg supplementation (500 mg/day) exacerbated colonic inflammation and fibrosis in rats because of excessive nitric oxide production and collagen deposition35. A study in humans36 reported that serum levels of L-Arg were 150% higher in patients with ulcerative colitis than in healthy individuals; levels of ornithine and lysine, which are also transported by CATs, were elevated as well, leading to competitive inhibition of arginine uptake. In the present study, AS II increased cellular L-Arg uptake without L-Arg supplementation. These findings suggest that enhancement of cellular L-Arg uptake, rather than L-Arg supplementation, may be a better strategy for treating IBD. L-glutamine supplementation has been shown to contribute to beneficial effects in a dextran sulfate sodium-induced colitis model37. The effect of AS II on L-glutamine absorption warrants further study.
L-Arg-mediated activation of the mTOR pathway improves intestinal cell migration and epithelial wound healing14, and was confirmed in genetically defined mouse models38. We found that AS II activated mTOR and its downstream targets, S6K and 4E-BP1. AS II increased protein synthesis in a concentration- and time-dependent manner, contributing to accelerated cell proliferation and wound healing. We confirmed the findings using lysine, a competitive inhibitor of L-arg uptake and rapamycin, a specific mTOR inhibitor. The mTOR signaling pathway is activated by several amino acids, including L-Arg and L-leucine39,40. Future studies should also assess the effect of AS II on other amino acids, such as L-leucine. In addition, the CATs protein levels may not be equal to the activity transport capacity. Our results show L-Arg uptake peaks at 6 h that are consistent with the previous study that the protein kinase C (PKC) activator 12-O-tetradecanoylphorbol 13-acetate (TPA), stimulated system y + arginine transport activity in Caco-2 cells with transport capacity (Vmax) between 6–12 h34. In addition, absorption of cationic amino acids is largely dependent on y+ transport system, such as CAT1 and CAT241. It suggests AS II might promote the activity of CATs via the PKC pathway. However, the relationship between CATs levels and activity requires further investigation.
In conclusion, AS II promotes intestinal epithelial repair by enhancing L-Arg uptake and activating the mTOR pathway. These findings suggest that AS II may be effective in relieving colitis.
Methods
Materials
A. membranaceus var. mongholicus was authenticated by Dr. H.C. Lin at the National Defense Medical Center, where a voucher specimen (NDMCP no. 1000901) has been deposited. L-Arg, L-lysine (Sigma-Aldrich, St. Louis, MO, USA), L-[3H]-Arg, L-[3H]-leucine (American Radiolabeled Chemicals, St. Louis, MO, USA), and Ultima Gold liquid scintillation cocktails (PerkinElmer, Waltham, MA, USA) were used in the study.
Preparation of AS II
Dried root powder of Radix Astragali (9.5 kg) was soaked in 95% ethanol (20 L × 7), yielding a 756.25-g solution of Radix Astragali extract with evaporation under reduced pressure. The extract was then partitioned between n-BuOH-H2O and n-hexane-90% MeOH to yield 90% MeOH layer (334.68 g). The 90% MeOH fraction was subjected to medium-pressure liquid chromatography and eluted with an H2O-MeOH gradient system, yielding three fractions. Fraction 2 (83.93 g) was further chromatographed on silica gel and eluted with CHCl3-MeOH-H2O (10:5:1) to yield AS II (1.68 g), which was identified using spectral data in the literature (Fig. 1A)42,43.
Cell culture
The human intestinal epithelial cell line Caco-2, a widely used model for studying the intestinal barrier, permeability, and wound healing44, was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) with regular supplements.
Scratch wound assay
The assay was performed as described previously45. Briefly, Caco-2 cells were seeded in 24-well plates (2 × 105 cells/well) and allowed to reach confluence. Scratch wounds were made using a sterile 10-μL pipette tip to create a straight cell-free line simulating a wound. After scratching, the cells were rinsed gently with phosphate-buffered saline (PBS) to remove detached cells. Next, cells were incubated with AS II in 5% CO2 at 37 °C. Cell migration was measured using photomicrography equipment (Leica Microsystems, Wetzlar, Germany) to compare the wound area 0, 12, 24, 36, and 48 hr after making the scratch.
Cell proliferation assay
Cell viability was assessed using a cell counting kit (CCK-8, Dojindo, Kumamoto, Japan) as described previously46. Briefly, Caco-2 cells (5 × 103 cells/well) were seeded in 96-well plates and incubated in serum-free medium with various concentrations of AS II for 48 hr. The medium was then removed, and 10 μL of CCK-8 in 90 μL of PBS were added to the cells for 2 hr. Absorbance was measured at 450 nm using a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
L-Arg uptake assay
The assay was carried out as described previously11. Briefly, Caco-2 cells (2.5 × 104 cells/well) were seeded in 24-well plates. Cells were cultured for 7 days after reaching confluence to allow differentiation. The cells were then rinsed three times with transport buffer and incubated in transport buffer containing 0.1 mM L-Arg and 1 μCi/mL L-[3H]-Arg for 5 min. The buffer was then removed; cells were rinsed three times with cold PBS and dissolved in 300 μL lysis buffer (1 N NaOH) for 1 hr at room temperature. Tissue L-Arg uptake measurement was performed as described previously7. A 1.5-cm section of intestinal or colonic tissue was dissected and immediately incubated in transport buffer containing 0.1 mM L-Arg and 1 μCi/mL L-[3H]-Arg for 5 min. The transport buffer was then removed; the tissues were rinsed three times with cold PBS and then lysed in 500 μL 0.1 NHNO3 by gently shaking for 24 hr at room temperature. A 200-μL aliquot of the lysate was then collected and mixed with 2 mL of liquid scintillation cocktails. Radioactivity was measured with a scintillation counter (TopCount, Packard BioScience, Meriden, CT, USA). L-Arg uptake is expressed as nmol/mg of protein/min.
De novo protein synthesis assay
The assay was described in a previous study47. Briefly, Caco-2 cells were seeded in 24-well plates (5 × 104 cells/well) and cultured for 7 days after reaching confluence. Cells were incubated in DMEM containing 1 μCi/ml [3H]-leucine for 4 hr and then rinsed three times with cold PBS. Next, cells were dissolved in 200 μL lysis buffer and precipitated with 10% trichloroacetic acid (TCA) for 10 min. After centrifugation at 12,000 g for 5 min, the supernatant was discarded and the protein pellet was rinsed with 10% TCA and then dissolved in 300 μL NaOH (1 N). The solution (200 μL) was mixed with 2 mL of liquid scintillation cocktails. Protein synthesis was measured with a scintillation counter (TopCount, Packard BioScience).
Animal study protocol
Male C57BL/6JNarl 5-week-old (18–22 g) mice were purchased from the National Laboratory Animal Center (Taipei, Taiwan). Animal feeding and experimental procedures were approved by the Institutional Animal Care and Use Committee of the National Defense Medical Center (certificate number: IACUC-13–302) and performed in accordance with the relevant guidelines and regulations48. The mice were gavaged with 1 mL saline or AS II (10 mg/kg) once daily for 11 consecutive days. TNBS (100 mg/kg) was administered via the rectum on the 8th day49. The mice were anesthetized with pentobarbital sodium (i.p. 70 mg/kg), and blood samples were collected from the orbital sinus. At the end of the experiment (11th day), the animals were euthanized using CO2 to allow harvesting of tissues. The intestine and colon were dissected.
MPO activity in colon tissue
MPO activity was assayed as previously described50. In brief, the tissue was lysed and freeze-thawed for three cycles in extraction buffer (1:20, w/v). Homogenates were then centrifuged at 14,000 rpm for 20 min. Ten microliters of supernatant were collected and mixed with 190 μL assay buffer (1.68 mM 3,3′,5,5′-tetramethylbenzidine and 0.00015% hydrogen peroxide). MPO activity was determined at 650 nm using a microplate reader (Molecular Devices).
Intestinal permeability assay
The assay was performed as described previously51. Briefly, the mice were fasted for 8 hr and then gavaged with FITC-dextran (50 mg/100 g; Sigma-Aldrich) 4 hr after fasting. Blood samples were collected from the retro-orbital sinus and centrifuged at 12,000 g for 20 min. Serum was measured at excitation and emission wavelengths of 490 and 520 nm, respectively, using a fluorescence microtiter plate reader (POLARstar Galaxy; BMG Labtech, Ortenberg, Germany).
Western blot analysis
This method was described in our previous study48. Cells were plated in 6-well plates (1 × 106 cells/well) and treated accordingly. Cells were then harvested in 0.2 mL of RIPA lysis buffer, separated by 10% SDS-PAGE, and transferred to PVDF membranes (Millipore, Billerica, MA, USA). Immunoblotting was performed using primary antibodies against CAT1, CAT2 (Abcam, Cambridge, UK), p-mTOR, mTOR, p-p70S6K, p70S6K, p-4E-BP1, 4E-BP1 (Cell Signaling Technology, Beverly, MA, USA), and the housekeeper β-actin (Millipore). The signals were visualized with an enhanced chemoluminescence kit (Amersham Biosciences, Little Chalfont, UK) followed by exposure of the blots to X-ray film.
Histology
A histological examination was performed following a method described previously50. Intestinal tissues were soaked in 10% formaldehyde solution for 24 hr, then stained with hematoxylin and eosin.
Statistical analysis
All data represent the mean ± SEM. Significant differences between group means were determined by one-way ANOVA followed by a Bonferroni post hoc test using SPSS version 22 (IBM SPSS, Armonk, NY, USA). p < 0.05 was considered statistically significant.
References
Xavier, R. J. & Podolsky, D. K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434, https://doi.org/10.1038/nature06005 (2007).
Wilkins, T., Jarvis, K. & Patel, J. Diagnosis and Management of Crohn’s Disease. Am Fam Physician 84, 1365–1375 (2011).
Zaki, M. H., Lamkanfi, M. & Kanneganti, T. D. The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends in immunology 32, 171–179, https://doi.org/10.1016/j.it.2011.02.002 (2011).
Iskandar, H. N. & Ciorba, M. A. Biomarkers in inflammatory bowel disease: current practices and recent advances. Translational research: the journal of laboratory and clinical medicine 159, 313–325, https://doi.org/10.1016/j.trsl.2012.01.001 (2012).
Porras, M. et al. Correlation between cyclical epithelial barrier dysfunction and bacterial translocation in the relapses of intestinal inflammation. Inflammatory bowel diseases 12, 843–852, https://doi.org/10.1097/01.mib.0000231571.88806.62 (2006).
Terc, J., Hansen, A., Alston, L. & Hirota, S. A. Pregnane X receptor agonists enhance intestinal epithelial wound healing and repair of the intestinal barrier following the induction of experimental colitis. European journal of pharmaceutical sciences: official journal of the European Federation for Pharmaceutical Sciences 55, 12–19, https://doi.org/10.1016/j.ejps.2014.01.007 (2014).
Coburn, L. A. et al. L-arginine supplementation improves responses to injury and inflammation in dextran sulfate sodium colitis. PloS one 7, e33546, https://doi.org/10.1371/journal.pone.0033546 (2012).
Catalioto, R. M., Maggi, C. A. & Giuliani, S. Intestinal epithelial barrier dysfunction in disease and possible therapeutical interventions. Current medicinal chemistry 18, 398–426 (2011).
Maloy, K. J. & Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306, https://doi.org/10.1038/nature10208 (2011).
Singh, K. et al. L-arginine uptake by cationic amino acid transporter 2 is essential for colonic epithelial cell restitution. American journal of physiology. Gastrointestinal and liver physiology 302, G1061–1073, https://doi.org/10.1152/ajpgi.00544.2011 (2012).
Visigalli, R. et al. Regulation of arginine transport and metabolism by protein kinase Calpha in endothelial cells: stimulation of CAT2 transporters and arginase activity. Journal of molecular and cellular cardiology 49, 260–270, https://doi.org/10.1016/j.yjmcc.2010.04.007 (2010).
Stechmiller, J. K., Childress, B. & Cowan, L. Arginine supplementation and wound healing. Nutrition in clinical practice: official publication of the American Society for Parenteral and Enteral Nutrition 20, 52–61 (2005).
Bertrand, J., Goichon, A., Dechelotte, P. & Coeffier, M. Regulation of intestinal protein metabolism by amino acids. Amino acids 45, 443–450, https://doi.org/10.1007/s00726-012-1325-8 (2013).
Bauchart-Thevret, C., Cui, L., Wu, G. & Burrin, D. G. Arginine-induced stimulation of protein synthesis and survival in IPEC-J2 cells is mediated by mTOR but not nitric oxide. American journal of physiology. Endocrinology and metabolism 299, E899–909, https://doi.org/10.1152/ajpendo.00068.2010 (2010).
Kim, E. Mechanisms of amino acid sensing in mTOR signaling pathway. Nutrition research and practice 3, 64–71, https://doi.org/10.4162/nrp.2009.3.1.64 (2009).
Rhoads, J. M., Niu, X., Odle, J. & Graves, L. M. Role of mTOR signaling in intestinal cell migration. American journal of physiology. Gastrointestinal and liver physiology 291, G510–517, https://doi.org/10.1152/ajpgi.00189.2005 (2006).
Liu, J., Zhao, Z. Z. & Chen, H. B. Review of Astragali Radix. Chinese Herbal Medicines 3, 90–105, https://doi.org/10.3969/j.issn.1674-6384.2011.02.004 (2011).
Xu, Q., Ma, X. & Liang, X. Determination of astragalosides in the roots of Astragalus spp. using liquid chromatography tandem atmospheric pressure chemical ionization mass spectrometry. Phytochemical analysis: PCA 18, 419–427, https://doi.org/10.1002/pca.997 (2007).
Huang, C. et al. Reversal of P-glycoprotein-mediated multidrug resistance of human hepatic cancer cells by Astragaloside II. The Journal of pharmacy and pharmacology 64, 1741–1750, https://doi.org/10.1111/j.2042-7158.2012.01549.x (2012).
Kong, X. H. et al. Astragaloside II induces osteogenic activities of osteoblasts through the bone morphogenetic protein-2/MAPK and Smad1/5/8 pathways. International journal of molecular medicine 29, 1090–1098, https://doi.org/10.3892/ijmm.2012.941 (2012).
Wan, C. P. et al. Astragaloside II triggers T cell activation through regulation of CD45 protein tyrosine phosphatase activity. Acta pharmacologica Sinica 34, 522–530, https://doi.org/10.1038/aps.2012.208 (2013).
Vuksan, V. et al. American ginseng (Panax quinquefolius L.) attenuates postprandial glycemia in a time-dependent but not dose-dependent manner in healthy individuals. The American journal of clinical nutrition 73, 753–758 (2001).
Iacucci, M. & Ghosh, S. Looking beyond symptom relief: evolution of mucosal healing in inflammatory bowel disease. Therapeutic advances in gastroenterology 4, 129–143, https://doi.org/10.1177/1756283x11398930 (2011).
Zhao, L. et al. The in vivo and in vitro study of polysaccharides from a two-herb formula on ulcerative colitis and potential mechanism of action. Journal of ethnopharmacology 153, 151–159, https://doi.org/10.1016/j.jep.2014.02.008 (2014).
Ko, J. K., Lam, F. Y. & Cheung, A. P. Amelioration of experimental colitis by Astragalus membranaceus through anti-oxidation and inhibition of adhesion molecule synthesis. World journal of gastroenterology: WJG 11, 5787–5794 (2005).
Ko, J. K. & Chik, C. W. The protective action of radix Astragalus membranaceus against hapten-induced colitis through modulation of cytokines. Cytokine 47, 85–90, https://doi.org/10.1016/j.cyto.2009.05.014 (2009).
Sevimli-Gur, C. et al. In vitro growth stimulatory and in vivo wound healing studies on cycloartane-type saponins of Astragalus genus. Journal of ethnopharmacology 134, 844–850, https://doi.org/10.1016/j.jep.2011.01.030 (2011).
Shan, Y. H. et al. Silk fibroin/gelatin electrospun nanofibrous dressing functionalized with astragaloside IV induces healing and anti-scar effects on burn wound. International journal of pharmaceutics 479, 291–301, https://doi.org/10.1016/j.ijpharm.2014.12.067 (2015).
Triantafillidis, J. K., Merikas, E. & Georgopoulos, F. Current and emerging drugs for the treatment of inflammatory bowel disease. Drug design, development and therapy 5, 185–210, https://doi.org/10.2147/DDDT.S11290 (2011).
Lee, Kuo-Hsiung et al. Recent Progress of Research on Herbal Products Used in Traditional Chinese Medicine: the Herbs belonging to The Divine Husbandman’s Herbal Foundation Canon (神農本草經 Shén Nóng Běn Cǎo Jīng). J Tradit Complement Med 2, 6–26, https://doi.org/10.1269/jrr.08066 (2012).
Dong, J. et al. Effects of large dose of Astragalus membranaceus on the dendritic cell induction of peripheral mononuclear cell and antigen presenting ability of dendritic cells in children with acute leukemia. Zhongguo Zhong Xi Yi Jie He Za Zhi 25, 872–875, https://doi.org/10.2147/DDDT.S11290 (2005).
Witte, M. B. & Barbul, A. Arginine physiology and its implication for wound healing. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society 11, 419–423 (2003).
Debats, I. B. et al. Role of arginine in superficial wound healing in man. Nitric oxide: biology and chemistry/official journal of the Nitric Oxide Society 21, 175–183, https://doi.org/10.1016/j.niox.2009.07.006 (2009).
Pan, M. & Stevens, B. R. Protein kinase C-dependent regulation of L-arginine transport activity in Caco-2 intestinal cells. Biochimica et biophysica acta 1239, 27–32 (1995).
Mane, J. et al. Effect of L-arginine on the course of experimental colitis. Clinical nutrition 20, 415–422, https://doi.org/10.1054/clnu.2001.0469 (2001).
Hong, S. K. et al. Increased serum levels of L-arginine in ulcerative colitis and correlation with disease severity. Inflammatory bowel diseases 16, 105–111, https://doi.org/10.1002/ibd.21035 (2010).
Ren, W. et al. Serum amino acids profile and the beneficial effects of L-arginine or L-glutamine supplementation in dextran sulfate sodium colitis. PloS one 9, e88335, https://doi.org/10.1371/journal.pone.0088335 (2014).
Squarize, C. H., Castilho, R. M., Bugge, T. H. & Gutkind, J. S. Accelerated wound healing by mTOR activation in genetically defined mouse models. PloS one 5, e10643, https://doi.org/10.1371/journal.pone.0010643 (2010).
Ban, H. et al. Arginine and Leucine regulate p70 S6 kinase and 4E-BP1 in intestinal epithelial cells. International journal of molecular medicine 13, 537–543 (2004).
Kim, J. et al. Arginine, leucine, and glutamine stimulate proliferation of porcine trophectoderm cells through the MTOR-RPS6K-RPS6-EIF4EBP1 signal transduction pathway. Biology of reproduction 88, 113, https://doi.org/10.1095/biolreprod.112.105080 (2013).
Singh, K. et al. L-arginine uptake by cationic amino acid transporter 2 is essential for colonic epithelial cell restitution. Am J Physiol Gastrointest Liver Physiol. 302, G1061–1073, https://doi.org/10.1152/ajpgi.00544.2011.-Restoration (2012).
Kitagawa, I., Wang, H. K., Saito, M., Takagi, A. & Yoshikawa, M. Saponin and sapogenol. XXXV. Chemical constituents of astragali radix, the root of Astragalus membranaceus Bunge.(2). Astragalosides I, II and IV, acetylastragaloside I and isoastragalosides I and II. Chemical & Pharmaceutical Bulletin 31, 698–708 (1983).
Kitagawa, I., Wang, H. K., Saito, M. & Yoshikawa, M. Saponin and Sapogenol.)(XXVI.1) Chemical Constituents of Astragali Radix, the Root of Astragalus membranaceus Bunge. (3). Astragalosides III, V and VI. Chem. Pharm. Bull. 31, 709–715 (1983).
Sambuy, Y. et al. The Caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell biology and toxicology 21, 1–26, https://doi.org/10.1007/s10565-005-0085-6 (2005).
Ranzato, E., Patrone, M., Mazzucco, L. & Burlando, B. Platelet lysate stimulates wound repair of HaCaT keratinocytes. The British journal of dermatology 159, 537–545, https://doi.org/10.1111/j.1365-2133.2008.08699.x (2008).
Lee, S. Y. et al. Rhodiola crenulata and Its Bioactive Components, Salidroside and Tyrosol, Reverse the Hypoxia-Induced Reduction of Plasma-Membrane-Associated Na,K-ATPase Expression via Inhibition of ROS-AMPK-PKCξ Pathway. Evidence-based complementary and alternative medicine: eCAM 2013, 1–15, https://doi.org/10.1155/2013/284150 (2013).
Oubrahim, H., Wong, A., Wilson, B. A. & Chock, P. B. Mammalian target of rapamycin complex 1 (mTORC1) plays a role in Pasteurella multocida toxin (PMT)-induced protein synthesis and proliferation in Swiss 3T3 cells. The Journal of biological chemistry 288, 2805–2815, https://doi.org/10.1074/jbc.M112.427351 (2013).
Lee, S.-Y. et al. Rhodiola crenulata extract suppresses hepatic gluconeogenesis via activation of the AMPK pathway. Phytomedicine: international journal of phytotherapy and phytopharmacology 22, 477–486, https://doi.org/10.1016/j.phymed.2015.01.016 (2015).
Scheiffele, F. & Fuss, I. J. Induction of TNBS colitis in mice. Current protocols in immunology/edited by John E. Coligan… [et al.] Chapter 15, Unit15.19, doi:https://doi.org/10.1002/0471142735.im1519s49 (2002).
Lee, S. Y. et al. Rhodiola crenulata Extract Alleviates Hypoxic Pulmonary Edema in Rats. Evidence-based complementary and alternative medicine: eCAM 2013, 718739, https://doi.org/10.1155/2013/718739 (2013).
Yan, Y. et al. Overexpression of Ste20-related proline/alanine-rich kinase exacerbates experimental colitis in mice. J Immunol 187, 1496–1505, https://doi.org/10.4049/jimmunol.1002910 (2011).
Acknowledgements
This work was supported by Grants from the Ministry of Science and Technology (MOST 104-2320-B-016-013 and MOST 105-2320-B-016-005 to T.-C.C. and MOST 104-2320-B-016-003 to S.-Y.L.) and the Ministry of National Defense (MAB-105-048 and MAB-106-071 to T.-C.C. and MAB-105-007 to S.-Y.L.), Taipei, Taiwan, ROC. We are also grateful to Nuliv Science, Inc., for generous assistance in this work.
Author information
Authors and Affiliations
Contributions
S.Y.L., W.C.T., and T.C.C. participated in the concept of the study and the experimental design. W.L.C. isolated and characterized the AS II compound. S.Y.L., W.C.T., and J.C.L. were involved in laboratory experiments and data analysis. S.F.H. contributed new reagents or analytic techniques. S.Y.L. wrote the manuscript. B.A.-S. and T.C.C. reviewed it.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, SY., Tsai, WC., Lin, JC. et al. Astragaloside II promotes intestinal epithelial repair by enhancing L-arginine uptake and activating the mTOR pathway. Sci Rep 7, 12302 (2017). https://doi.org/10.1038/s41598-017-12435-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12435-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
