
Abstract
Rare genomic copy number variations (CNVs) (frequency <1%) contribute a part to the genetic underpinnings of autism spectrum disorders (ASD). The study aimed to understand the scope of rare CNV in Taiwanese patients with ASD. We conducted a genome-wide CNV screening of 335 ASD patients (299 males, 36 females) from Taiwan using Affymetrix Genome-Wide Human SNP Array 6.0 and compared the incidence of rare CNV with that of 1093 control subjects (525 males, 568 females). We found a significantly increased global burden of rare CNVs in the ASD group compared to the controls as a whole or when the rare CNVs were classified by the size and types of CNV. Further analysis confirmed the presence of several rare CNVs at regions strongly associated with ASD as reported in the literature in our sample. Additionally, we detected several new private pathogenic CNVs in our samples and five patients carrying two pathogenic CNVs. Our data indicate that rare genomic CNVs contribute a part to the genetic landscape of our ASD patients. These CNVs are highly heterogeneous, and the clinical interpretation of the pathogenic CNVs of ASD is not straightforward in consideration of the incomplete penetrance, varied expressivity, and individual genetic background.
Similar content being viewed by others


Introduction
Autism spectrum disorder (ASD) represents a group of childhood-onset neurodevelopmental disorders characterized by abnormal social interactions, impaired verbal and nonverbal communication, and the presence of restricted interests and repetitive behaviors with long-term persistence of core features and functional impairment1,2. The prevalence of ASD is various across different regions with an increasing trend over the years3,4,5 and with male excess in a male-to-female ratio of approximately 5:16,7,8. In the USA, the prevalence of ASD increased in the past decade according to the report of Centers for Disease Control and Prevention of USA8. It was estimated that around 1 in 68 persons aged eight years in the USA in 2010 was affected with ASD8. However, the increasing trend of ASD prevalence was not observed in the UK9. The estimated prevalence of ASD in Chinese population ranged from 2.8 to 29.5 per 10,000 according to a recent review that summarized the findings in Chinese population from several areas3. The prevalence of ASD in Taiwan is approximately 0.3% based on the analysis of national health insurance research dataset10 and 1% based on the most recent Taiwan’s national survey of child and adolescent mental disorders11 with a male: female ratio of approximately 4: 110. Due to its high prevalence, long-term impairment resulting in a great impact on individuals, families, and society12,13 and strong evidence of genetic components in its etiology14, this severe developmental disorder has been prioritized for molecular genetic studies15.
The heritability estimate of ASD is greater than 90%, attesting that genetic factors play a major role in the pathogenesis of ASD16,17,18. However, the genetics of ASD is very complex. Several genome-wide association studies (GWAS) have identified some common single nucleotide polymorphisms (SNPs) associated with the risk of ASD, such as common variants on 20p12.1, 5p14.1, 1p13.219,20,21,22,23. These common SNPs, however, have only small effects on autism ASD risk, and of note, few if any of these SNPs were replicated in different studies. Furthermore, accumulating evidence suggests that rare genetic and genomic mutations also contribute to the genetics of ASD24. Conventional cytogenetic studies of ASD have revealed a variety of rare chromosomal abnormalities associated with ASD25,26,27,28 indicating aberrant genomic rearrangements are part of the genetic mechanism of ASD. Notably, the recent advent of array-based comparative genomic hybridization (aCGH) technology has discovered various submicroscopic copy number variations (CNVs) of genomic DNA associated with ASD29,30, leading further support to the idea that ASD is a genomic disorder in a subset of the patients. These ASD-associated CNVs are usually individually unique and of low frequency, but together they account for approximately 5–10% of idiopathic ASD29, hence, constituting a part of the genetic architecture of ASD22,31,32. The discovery of genomic mutations in ASD-associated CNVs not only helps decipher the genetic complexity of ASD23, but also helps shed some light on the neurobiology and pathogenesis of ASD32,33,34,35,36,37.
In our previous studies, we reported four pathogenic CNVs in certain ASD patients38,39, indicating that CNVs also play a role in the genetic architecture of ASD in our patients. To have a better understanding of the scope of rare genomic CNVs in our ASD patient population, we recruited a sample of more than 300 ASD patients and conducted a genome-wide CNV screening in this sample.
Results
Clinical characteristics
A total of 335 (95.7%) out of 350 cases and 1093 (98.4%) out of 1111 controls passed a series of quality control of CNV experiments. We investigated the ethnicity of cases and controls by performing principle component analysis (PCA) with SNP genotype data from all the participants of this study and the individuals included in HapMap study. The results demonstrated that the cases and controls are clustered together with the Han Chinese (Supplementary Figure 1). Therefore, the ethnicity of the participants of this study was confirmed to be the Han Chinese. Further, all the CNV data were subjected to the burden analysis. The patient group consisted of 299 boys and 36 girls with the mean age of 9.4 ± 4.0 years, while the control group consisted of 525 males and 568 females with the mean age of 68.1 ± 10.1 years. The ADI-R (Autism Diagnostic Interview-Revised) interviews revealed that the 335 patients scored 20.43 ± 6.12 in the “qualitative abnormalities in reciprocal social interaction”, 14.75 ± 4.32 in the “qualitative abnormalities in communication, verbal”, 8.19 ± 3.33 in the “qualitative abnormalities in communication, nonverbal”, and 6.95 ± 2.47 in the “restricted, repetitive and stereotyped patterns of behaviors.” All the participants with ASD were noted to have had abnormal development at or before 36 months of age. Their current average intelligence quotients (IQ) were 94.85 ± 22.55 (range, 40 to 148) for full-scale IQ, 96.74 ± 2.04 (range, 41 to 145) for performance IQ, and 95.08 ± 23.79 (range, 44 to 148) for verbal IQ. Among the 335 ASD patients, nine had been diagnosed with epilepsy (3.04%), four had been suspected of seizure (1.35%), and 19 had ever had a febrile convulsion (6.42%). These data are also provided in the Supplementary Table 1.
CNV findings
The rates of rare CNV (<1% in the patients) at autosomes and X-chromosome were examined between the patient and control groups. CNV regions on autosomes were analyzed in all samples while CNV regions on sex chromosomes were analyzed in male samples only. We found a significant excess of the overall rate of rare CNV at autosomes in the ASD patients (2.71) compared with the control subjects (0.77). The over-representation of rare autosomal CNV rate in ASD was still present when the rare CNVs were grouped into deletion and duplication or classified according to the size as <100 kb, 100–400 kb, and >400 kb (Table 1). In the analysis of rare CNV at X-chromosome, we compared only the rate of rare CNV between male patients and male control subjects. A significant excess of the overall rate of rare CNV was observed in the male ASD patients (0.214) compared with the male control subjects (0.011). The excess rate of rare CNV at X-chromosome was still present when the CNVs were stratified into deletion/duplication, or different size groups (Table 2). We did not compare the rate of rare CNV at X-chromosome between female patients and female controls, because of the random inactivation of X-chromosome in the females. Additionally, the sample size of the female patients is small (n = 36) compared to the female controls (n = 568), and the skewed proportion of female patients vs. female controls (0.11 vs. 0.52).
CNVs at “hot spots”
We compared the rare CNVs found in our patients with the selected genetic “hot spots” of ASD as reported in the paper entitled “Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions” from the practice guideline of the American College of Medical Genetics and Genomics40. The “hot spots” was defined as CNVs that have an especially strong association with ASD according to this paper. We identified a total of 14 patients who had pathogenic CNVs located at several of the “hot spots.” The detailed information of these CNVs including the locations, sizes, and genes encompassed in the CNV region are listed in Table 3, while the clinical data of each patient are listed in the Supplementary Table 2.
Other rare pathogenic CNVs
Besides the detection of CNVs at the “hot spots” in our sample, we further identified a total of 49 rare putative pathogenic CNVs according to the “American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants”41 in our sample. The pathogenic CNV was defined as “documented as clinically significant in multiple peer-reviewed publications, even if penetrance and expressivity of the CNV are known to be variable. This category includes large CNVs, which may not be described in the medical literature at the size observed in the patient but which overlap a smaller interval with clearly established clinical significance”41. These putative pathogenic CNVs overlapped with the pathogenic CNVs reported in the Clinical Genome Resources CNVs and DECIPHER. Table 4 presents the detailed information of these CNVs including locations, types, origins, and genes encompassed. The clinical data of each patient are provided in the Supplementary Table 3.
Two-hit CNVs
Five patients were found to have two different putative pathogenic CNVs simultaneously in this study. Patient U-2075 inherited the 4q amplification and the 5p deletion from his mother and father, respectively (Fig. 1A). Patient U-1753 had the 8q amplification and the 8p deletion transmitted from his mother and father, respectively (Fig. 1B). Patient U-1255 acquired the 10q amplification and 18p amplification from his mother and father, respectively (Fig. 1C). Patient U-1414 had the 8p amplification from his father and a de novo 9q duplication (Fig. 1D). Patient U-1999 had two de novo amplifications at 17q25.3 simultaneously (Fig. 1E). All the parents of these five patients were carefully assessed, and none of them had ASD based on the self-administered questionnaires and clinical evaluation by the corresponding author. The detailed information of these CNVs including the locations, sizes, and genes encompassed by these CNVs are listed in Table 5, and the clinical data of each patient are provided in the Supplementary Table 4.

The pedigrees of five patients who carry two CNVs and the origins of these CNVs. Dup: duplication, Del: deletion.
Discussion
In this study, we compared the frequencies of rare CNVs (<1%) between 335 patients with ASD and 1093 control subjects from Taiwan. We found a significantly higher frequency of global rare CNVs in patients with ASD compared to the control group. The significantly higher frequencies of rare CNVs in the ASD group were still present when the CNVs were subdivided into different groups based on deletion/duplication or the sizes. Our data are compatible with several previous studies24,33,42. Pinto and colleagues conducted a genome-wide CNV analysis of 996 ASD individuals of European ancestry and 1,287 matched controls. They found a higher global burden of rare genic CNVs in ASD patients43. The findings were replicated by the same group in another genome-wide CNV analysis consisted of 2,446 families with ASD33. In our study, we did not limit our CNV analysis to genic CNVs only, as non-genic CNVs may have position effect to affect the expression of genes outside the CNV regions. Our findings of increased global burden of rare CNVs in ASD indicate that genomic rearrangement is one of the genetic mechanisms of ASD.
Some other studies reported increased burden of CNV in female patients. Jacquemont and colleagues reported that in a sample of 762 ASD families, they found a 3-fold increase in deleterious autosomal CNVs in female patients compared to male probands44. Desachy and colleagues recently reported that mothers of patients with autism had a higher deletion burden than control mothers in a matched case–control population. Also, to their surprise, they found a higher autosomal burden of large, rare CNVs in females in the population. They speculated that the increased rare CNV burden in females in general population might contribute to the decreased female fetal loss in the population, but the ASD-specific maternal CNV burden may contribute to high sibling recurrence45. In our study, we did not conduct the similar analysis because of the relatively small sample size of female patients.
In our CNV analysis, we identified 14 patients who had CNVs located at the ASD “hot spots” as reported in the “Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions”40. Our findings support the strong association of the CNVs at “hot spots” with ASD in our patient population. Among these CNVs, CNVs located at 22q11.2, 22q13.3, and 15q11-13 are the most common in our sample. Besides the CNVs located at the “hot spots,” we also detected 49 rare (<1%) CNVs larger than 400 kb that overlapped with the pathogenic CNVs reported in the Clinical Genome Resources and DECIPHER. These CNVs met the criteria of “pathogenic” according to the “American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants”41. Some of these rare putative pathogenic CNVs were inherited, while some were de novo mutations. The broad distributions of both “hot spots” CNVs and rare pathogenic CNVs detected in our patients suggest extremely high genetic heterogeneity of ASD in our patients. Some studies suggested that female patients have a higher burden of CNV than male patients33,44,45. But, in our study, all the patients who had CNVs at “hot spots” were male (Table 3), and there were only two female patients out of 49 who had other rare (<1%) pathogenic CNVs (Table 4). The discrepancy might be due to the disproportion of female patients in this study (36 females vs. 299 males).
In this study, five patients were found to have the concomitant presence of two rare pathogenic CNVs in their genome. The finding is consistent with our previous report of a patient who had inherited two CNVs from his parents and supported the two-hit model of ASD39. Several studies also proposed the idea of two-hit and the multiple-hit models of ASD, suggesting that genetic underpinnings of ASD stem from combinatorial effects of mutations of oligogenic or multiple genes in different loci39,46,47,48. Leblond and colleagues reported three patients with deletions at SHANK2 gene locus. Also, these three patients had another inherited CNV at 15q11-13 that was associated with other psychiatric disorders46. Two patients carried a duplication of nicotinic receptor CHRNA7, and one patient had a deletion of the synaptic translation repressor CYFIP1 46. Stenberg and Webber conducted a pathway-association test of target genes regulated by fragile-X mental retardation protein (FMRP) in ASD patients; they found rigorous support for the multiple-hit genetic etiology of ASD47. In fact, emerging evidence suggests the presence of multiple pathogenic CNVs in psychiatric patients is not rare. Hu and colleagues recently reported a novel maternally inherited 8q24.3 and a rare paternally inherited 14q23.3 CNVs in a family with neurodevelopmental disorders49. Williams and colleagues conducted CNV analysis in patients with velo-cardio-facial syndrome (VCFS), regardless of having psychosis or not. They found a significantly higher proportion of second CNV hit in patients with psychosis, suggesting the two-hit hypothesis may be relevant to a proportion of VCFS patients with psychosis50. Rudd and colleagues found a slightly higher proportion of multiple conservative CNVs in schizophrenia patients compared to controls, indicating a potential role for a multiple-hit model in schizophrenia51. Hence, it is likely that the multiple-hit (including two-hit) might be a commonly important genetic mechanism associated with ASD. In this study, we reported 5 patients who had two putative pathogenic CNVs larger than 400 kb. We believe that if CNVs smaller than 400 Kb were included for analysis in the future, we might find more patients with two-hit and multiple-hit of CNVs. In the family study of these 5 patients with two-hit CNVs, we found that some of these putative pathogenic CNVs were inherited from their parents, and some were a de novo mutation. However, the parents who carried one putative pathogenic CNV did not manifest ASD symptoms after careful clinical evaluation, suggesting the incomplete penetrance of these inherited pathogenic CNVs. Further, we searched for these CNVs in 1093 control subjects, and found none of these CNVs in the control group, except the duplication of 18p11.31-p11.2 (483 kb), which was found in 5 out of 1093 control subjects (0.46%). These data provided further evidence to support that these CNVs may confer increased risk to ASD. In the future, we might be able to identify unaffected carriers of high-risk CNVs if more data are accumulated.
The identification of pathogenic CNVs associated with ASD may help discover candidate genes of ASD. The genes encompassed by the CNVs in our patients are listed in Tables 3,4 and 5. Notably, several genes had been reported to be associated with autism or the other major psychiatric disorders, such as TACR1 52, CNTNAP5 53,54, ADGRL3 55, ZNF827 56, POU6F2 20, KMT2E 57, KCND2 58, SND1 59, CNTNAP2 60,61, CSMD1 62,63, PARD3 64, HERC2 65, FMN1 66, and RBFOX1 67. These findings not only provide further clues to indicate the highly genetic heterogeneity of ASD but also indicate the pleiotropic clinical effects of the mutation of these genes. Accumulating evidence showed that shared heritability and genetic mutations among different categories of psychiatric disorders seem to be regular rather than exceptional. A study of analyzing the genotype data from the Psychiatric Genomics Consortium (PGC) for cases and controls in schizophrenia, bipolar disorder, major depressive disorder, ASD and attention-deficit/hyperactivity disorder (ADHD) revealed moderate to high shared genetic etiology of these psychiatric disorders68. Li and colleagues recently reported that the prevalence of de novo mutations shared by four different categories of neuropsychiatric disorders: autism spectrum disorder, epileptic encephalopathy, intellectual disability, and schizophrenia, was significantly elevated69.
The present study has several limitations. First, to the best of our knowledge, our study has the largest sample size of Chinese population compared to the other studies70,71. However, the relatively limited sample size of this study is an apparent limitation to have a more comprehensive picture of CNVs in our patient population, especially in consideration of the high heterogeneity of CNVs associated with ASD. Second, in this study, we only searched for pathogenic CNVs larger than 400 kb because larger CNVs are more likely to be pathogenic72. We understand that small CNVs can be pathogenic. Hence, further analysis of the CNVs smaller than 400 kb will discover more ASD-associated CNVs in our patients. Third, the phenotypical interpretation of pathogenic CNVs found in our study is not straightforward given the incomplete penetrance, varied expressivity and pleiotropic effects of pathogenic CNVs identified in our sample. Also, we cannot exclude the interaction of these CNVs with the genetic background and other yet to be identified genetic or genomic mutations in the affected patients.
In conclusion, we found a significantly increased global burden of rare CNVs in our ASD patients compared to the control subjects, indicating that rare CNVs play a part in the genetic landscape of ASD in our population. Also, we identified several pathogenic CNVs at “hot spots” and various private putative pathogenic CNVs in our patients, suggesting high genetic heterogeneity of ASD in our patients. Our study also supports that high-resolution oligonucleotide SNP array is a useful tool to uncover the genetic underpinnings of patients with ASD. In the future, we will continue to analyze our data with the size of CNV smaller than <400 kb, and we expect to find more pathogenic CNVs associated with ASD, more candidate genes of ASD, and more patients with double-hit of CNV. Thus, we will have a better understanding of the genetic architecture of ASD in our population.
Materials and Methods
Participants and Procedures
The study protocol was approved by the Research Ethics Committee at National Taiwan University Hospital (approval number: 9561709027), Taipei, Taiwan, and Chang Gung Memorial Hospital-Linkou (approval number: 93-6244), Taiwan, for the recruitment of the patients with ASD, and Academia Sinica (approval number, AS-IBMS-MREC-91-10), Taiwan for the control group. All the experiments and informed consent procedures were performed in accordance with relevant guidelines and regulations set by the research ethics committees of the three institutes.
Patient Participants with ASD
The study was part of the molecular genetics study of patients with ASD who were Han Chinese residing in Taiwan. The detailed recruitment and evaluation of the patients and their family members were described in our previous publication73. In brief, patients aged 3 to 17 years old and met the clinical diagnosis of autistic disorder as defined by the Diagnostic and Statistical Manual of Mental Disorders-IV (DSM-IV)74 were recruited from the Children’s Mental Health Center, National Taiwan University Hospital, Taipei, Taiwan and Department of Psychiatry, Chang-Gung Memorial Hospital, Kuei-Shan, Taiwan. The clinical diagnoses were made by board-certified child psychiatrists experienced in the assessment and intervention for ASD and were further confirmed by interviewing the parents using the Chinese version of the Autism Diagnostic Interview-Revised (ADI-R)75,76. The ADI-R, translated into Mandarin by Gau and colleagues, was approved by Western Psychological Services in 2007 as the ADI-R in the Chinese language75,77. All these patient participants further received clinical evaluation according to the DSM-5 diagnostic criteria for ASD1, which revealed that all the 350 participants with DSM-IV autistic disorder met the diagnosis of DSM-5 ASD. Moreover, all these patient participants received intelligence tests. For ages of 3 to 7.5 years, the Wechsler Primary and Preschool Scale of Intelligence-Revised (WPPSI-R) was given; for ages of 6 to 16 years 11 months, the Wechsler Intelligence Scale for Children-3rd Edition (WISC-III) was given; for ages of 16 years and above, Wechsler Adult Intelligence Scale (WAIS) was given.
Patients with known chromosomal abnormalities and associated medical conditions including fragile X syndrome and Rett’s disorder based on DNA testing or clinical assessments were not included during the recruitment process28,78. Also, probands with previously identified chromosomal structural abnormality associated with autism or had any other major neurological or medical conditions were also excluded28. The study protocol was approved by the Research Ethics Committee of National Taiwan University Hospital (approval number, 9561709027) and Chang Gung Memorial Hospital (approval number, 93-6244), Taiwan. Written informed consents were obtained from the participants (if applicable, otherwise, child assent) and their parents after the procedures were fully explained. Genomic DNA was prepared from peripheral blood of each participant using Gentra Puregene Blood kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany).
Healthy control subjects
The control subjects (n = 1111) were chosen from the Han Chinese Cell and Genome Bank (HCCGB) in Taiwan who received physical check-up and questionnaire screening to ensure that they did not have any abnormal physical condition and mental illness79. Written informed consents were obtained from the participants after the procedures were fully explained.
CNV analysis
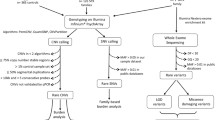
We used Affymetrix Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA, USA) for genome-wide CNV screening. The SNP 6.0 array contains more than 1.8 million markers including more than 906,600 probes for SNPs and more than 946,000 probes for CNVs. These probes are evenly distributed across the whole genome with a median distance between probes of ~0.7 kb. The microarray experiment was conducted by the National Genotyping Center (Academia Sinica, Taipei, Taiwan) (http://ncgm.sinica.edu.tw/ncgm_02/index.html). The hybridization intensities were captured by GeneChip Scanner 3000 (Affymetrix, Santa Clara, CA). CNVs were called using Affymetrix Genotyping Console software v.4.1 (Affymetrix, Santa Clara, CA). The average call rate was 99.49 ± 0.29%, and all samples passed genotyping quality control (call rate >= 95%). The gender was called based on the cn-probe-chrXY-ratio_gender method from Affymetrix Power Tools (Affymetrix, CA, USA). Samples with mismatched gender between computed gender and case information were excluded from analysis. Duplicated samples detected by Kinship analysis using P-Link software were also excluded. Twenty contiguous deletion or duplication probe signals were called in this study. CNV regions overlapped with centromeric regions (hg19, UCSC), antibody variable regions (PennCNV, http://www.openbioinformatics.org/penncnv/penncnv_faq.html#ig) and T-cell receptor loci (NCBI Gene, http://www.ncbi.nlm.nih.gov/gene/) were filtered out. The CNVs with the size equal or larger than 10 Kb and with the frequency of less than 1% in the patients were selected for analysis in this study. Copy number variations were considered to localize at the same locus if they overlapped by at least 80% of their length. Genes overlapped with the CNV regions were reported according to UCSC genes (NCBI37/hg19). The ethnicity of cases and controls was assessed by performing principle component analysis (PCA) with SNP genotype data from all the participants of this study and the individuals included in HapMap study.
Burden assay
Both genic and non-genic CNVs were included for analysis. Likelihood Ratio Chi-square test was used to compare the difference of CNV rate between ASD and healthy controls with a pre-selected alpha value at P value less than 0.05. Bonferroni correction was used to adjust for the multiple testing. Thus, the significance level of the p-value was set at 0.005.
Real-time quantitative PCR (RT-qPCR)
RT-qPCR was used to validate the CNVs detected in this study and for a family study to identify their parental origin. RT-qPCR was performed using the SYBR-Green PCR reagents kit (Applied Biosystems, Forster City, California, USA), and the CNV was assessed using a relatively standard method in the laboratory38. The experiment was implemented using the ABI StepOnePlus following the manufacturer’s protocol (Applied Biosystems, Forster City, California, USA). The description of primer sequences, optimal annealing temperature, and the amplicon sizes is available upon request.
Pathogenic CNV evaluation
The pathogenic CNVs were evaluated according to two practice guidelines from the American College of Genetics and Genomics. First, according to the “Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions”40, our CNVs results overlapped with those at the “hot spots” reported in this guideline are considered as pathogenic. Second, for rare CNVs outside the “hot spots,” we focused on the analysis of rare CNVs equal or larger than 400 kb for convenience’s sake. Although large CNVs are more likely to have clinical significance, we understand that small CNV can be pathogenic, and large CNV can be benign. CNVs overlapped with the pathogenic CNVs reported in the Clinical Genome Resources (https://www.clinicalgenome.org/), or DECIPHER (https://decipher.sanger.ac.uk/) were defined as pathogenic according to the “American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants”41. The sizes of CNVs and genes encompassed by the CNVs were generated according to the gene annotation of the UCSC genome browser (GRCh37/hg 19) (http://genome.ucsc.edu/cgi-bin/hgGateway).
References
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition 5th edn, (American Psychiatric Association 2013).
Lai, M. C., Lombardo, M. V. & Baron-Cohen, S. Autism. Lancet. 383, 896–910 (2014).
Feng, L. et al. Autism spectrum disorder in Chinese populations: a brief review. Asia Pac Psychiatry. 5, 54–60 (2013).
Isaksen, J., Diseth, T. H., Schjolberg, S. & Skjeldal, O. H. Autism spectrum disorders–are they really epidemic? Eur J Paediatr Neurol. 17, 327–333 (2013).
Kim, Y. S. et al. Prevalence of autism spectrum disorders in a total population sample. Am J Psychiatry 168, 904–912 (2011).
Ganz, M. L. The lifetime distribution of the incremental societal costs of autism. Arch Pediatr Adolesc Med. 161, 343–349 (2007).
Fombonne, E. Epidemiological surveys of autism and other pervasive developmental disorders: an update. J Autism Dev Disord. 33, 365–382 (2003).
CDC. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 63, 1–21 (2014).
Taylor, B., Jick, H. & Maclaughlin, D. Prevalence and incidence rates of autism in the UK: time trend from 2004-2010 in children aged 8 years. BMJ Open. 3, e003219, https://doi.org/10.1136/bmjopen-2013-003219 (2013).
Chien, I. C., Lin, C. H., Chou, Y. J. & Chou, P. Prevalence and incidence of autism spectrum disorders among national health insurance enrollees in Taiwan from 1996 to 2005. J Child Neurol. 26, 830–834 (2011).
Chen, Y. L. & Gau, S. S. The Mandarin version of the Kiddie-Schedule for Affective Disorders and Schizophrenia-Epidemiological Version for DSM-5- A psychometric study. J Formos Med Assoc (accepted) (2017).
Gau, S. S.-F. et al. Parental adjustment, marital relationship, and family function in families of children with autism. Res Autism Spectr Disord. 6, 263–270 (2012).
Buescher, A. V., Cidav, Z., Knapp, M. & Mandell, D. S. Costs of autism spectrum disorders in the United Kingdom and the United States. JAMA Pediatr. 168, 721–728 (2014).
Veenstra-Vanderweele, J., Christian, S. L. & Cook, E. H. Jr. Autism as a paradigmatic complex genetic disorder. Annu Rev Genomics Hum Genet. 5, 379–405 (2004).
Merikangas, K. R. & Risch, N. Genomic priorities and public health. Science. 302, 599–601 (2003).
Liu, J. et al. A genomewide screen for autism susceptibility loci. Am J Hum Genet. 69, 327–340 (2001).
Bailey, A. et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 25, 63–77 (1995).
Fombonne, E. Epidemiology of autistic disorder and other pervasive developmental disorders. J Clin Psychiatry. 66(Suppl 10), 3–8 (2005).
Anney, R. et al. Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum Mol Genet. 21, 4781–4792 (2012).
Anney, R. et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 19, 4072–4082 (2010).
Wang, K. et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 459, 528–533 (2009).
Xia, K. et al. Common genetic variants on 1p13.2 associate with risk of autism. Mol Psychiatry. 19, 1212–1219 (2014).
Bodmer, W. & Bonilla, C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 40, 695–701 (2008).
Liu, X. & Takumi, T. Genomic and genetic aspects of autism spectrum disorder. Biochem Biophys Res Commun. 452, 244–253 (2014).
Vorstman, J. A. et al. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry. 11, 18–28 (2006).
Margari, L., Lamanna, A. L., Craig, F. & Simone, M. & Gentile, M. Autism spectrum disorders in XYY syndrome: two new cases and systematic review of the literature. Eur J Pediatr. 173, 277–283 (2014).
Marshall, C. R. et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 82, 477–488 (2008).
Liao, H. M. et al. Chromosomal abnormalities in patients with autism spectrum disorders from Taiwan. Am J Med Genet B Neuropsychiatr Genet. 162B, 734–741 (2013).
Devlin, B. & Scherer, S. W. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev. 22, 229–237 (2012).
Rosti, R. O., Sadek, A. A., Vaux, K. K. & Gleeson, J. G. The genetic landscape of autism spectrum disorders. Dev Med Child Neurol. 56, 12–18 (2014).
Chung, B. H., Tao, V. Q. & Tso, W. W. Copy number variation and autism: new insights and clinical implications. J Formos Med Assoc. 113, 400–408 (2014).
Prasad, A. et al. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 (Bethesda). 2, 1665–1685 (2012).
Pinto, D. et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 94, 677–694 (2014).
Egger, G. et al. Identification of risk genes for autism spectrum disorder through copy number variation analysis in Austrian families. Neurogenetics. 15, 117–127 (2014).
Nomura, J. & Takumi, T. Animal models of psychiatric disorders that reflect human copy number variation. Neural Plast. 2012, 589524 (2012).
Luo, R. et al. Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am J Hum Genet. 91, 38–55 (2012).
Griswold, A. J. et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum Mol Genet. 21, 3513–3523 (2012).
Chien, W. H. et al. Identification and molecular characterization of two novel chromosomal deletions associated with autism. Clin Genet. 78, 449–456 (2010).
Gau, S. S., Liao, H. M., Hong, C. C., Chien, W. H. & Chen, C. H. Identification of two inherited copy number variants in a male with autism supports two-hit and compound heterozygosity models of autism. Am J Med Genet B Neuropsychiatr Genet. 159B, 710–717 (2012).
Schaefer, G. B., Mendelsohn, N. J. & Professional, P. & Guidelines, C. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet Med. 15, 399–407 (2013).
Kearney, H. M. et al. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 13, 680–685 (2011).
Kanduri, C. et al. The landscape of copy number variations in Finnish families with autism spectrum disorders. Autism Res. 9, 9–16 (2016).
Pinto, D. et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 466, 368–372 (2010).
Jacquemont, S. et al. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am J Hum Genet. 94, 415–425 (2014).
Desachy, G. et al. Increased female autosomal burden of rare copy number variants in human populations and in autism families. Mol Psychiatry. 20, 170–175 (2015).
Leblond, C. S. et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 8, e1002521, https://doi.org/10.1371/journal.pgen.1002521 (2012).
Steinberg, J. & Webber, C. The roles of FMRP-regulated genes in autism spectrum disorder: single- and multiple-hit genetic etiologies. Am J Hum Genet. 93, 825–839 (2013).
Rucker, J. J. & McGuffin, P. Polygenic heterogeneity: a complex model of genetic inheritance in psychiatric disorders. Biol Psychiatry. 68, 312–313 (2010).
Hu, J. et al. A novel maternally inherited 8q24.3 and a rare paternally inherited 14q23.3 CNVs in a family with neurodevelopmental disorders. Am J Med Genet A. 167A, 1921–1926 (2015).
Williams, H. J. et al. Schizophrenia two-hit hypothesis in velo-cardio facial syndrome. Am J Med Genet B Neuropsychiatr Genet. 162B, 177–182 (2013).
Rudd, D. S., Axelsen, M., Epping, E. A., Andreasen, N. C. & Wassink, T. H. A genome-wide CNV analysis of schizophrenia reveals a potential role for a multiple-hit model. Am J Med Genet B Neuropsychiatr Genet. 165B, 619–626 (2014).
Sharp, S. I. et al. Genetic association of the tachykinin receptor 1 TACR1 gene in bipolar disorder, attention deficit hyperactivity disorder, and the alcohol dependence syndrome. Am J Med Genet B Neuropsychiatr Genet. 165B, 373–380 (2014).
Pagnamenta, A. T. et al. Characterization of a family with rare deletions in CNTNAP5 and DOCK4 suggests novel risk loci for autism and dyslexia. Biol Psychiatry. 68, 320–328 (2010).
Djurovic, S. et al. A genome-wide association study of bipolar disorder in Norwegian individuals, followed by replication in Icelandic sample. J Affect Disord. 126, 312–316 (2010).
Arcos-Burgos, M. et al. A common variant of the latrophilin 3 gene, LPHN3, confers susceptibility to ADHD and predicts effectiveness of stimulant medication. Mol Psychiatry. 15, 1053–1066 (2010).
Jiang, S., Yang, W., Qiu, Y., Chen, H. Z. & Alzheimer’s Disease Neuroimaging Initiative. Identification of Novel Quantitative Traits-Associated Susceptibility Loci for APOE epsilon 4 Non-Carriers of Alzheimer’s Disease. Curr Alzheimer Res. 12, 218–227 (2015).
Dong, S. et al. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep. 9, 16–23 (2014).
Lee, H., Lin, M. C., Kornblum, H. I., Papazian, D. M. & Nelson, S. F. Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum Mol Genet. 23, 3481–3489 (2014).
Holt, R. et al. Linkage and candidate gene studies of autism spectrum disorders in European populations. Eur J Hum Genet. 18, 1013–1019 (2010).
Ji, W. et al. CNTNAP2 is significantly associated with schizophrenia and major depression in the Han Chinese population. Psychiatry Res. 207, 225–228 (2013).
Friedman, J. I. et al. CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol Psychiatry. 13, 261–266 (2008).
Havik, B. et al. The complement control-related genes CSMD1 and CSMD2 associate to schizophrenia. Biol Psychiatry 70, 35–42 (2011).
Xu, W. et al. Genome-wide association study of bipolar disorder in Canadian and UK populations corroborates disease loci including SYNE1 and CSMD1. BMC Med Genet. 15, 2 (2014).
Kim, S. K., Lee, J. Y., Park, H. J., Kim, J. W. & Chung, J. H. Association study between polymorphisms of the PARD3 gene and schizophrenia. Exp Ther Med. 3, 881–885 (2012).
Puffenberger, E. G. et al. A homozygous missense mutation in HERC2 associated with global developmental delay and autism spectrum disorder. Hum Mutat. 33, 1639–1646 (2012).
Cappi, C. et al. An inherited small microdeletion at 15q13.3 in a patient with early-onset obsessive-compulsive disorder. PloS One. 9, e110198, https://doi.org/10.1371/journal.pone.0110198 (2014).
Bill, B. R., Lowe, J. K., Dybuncio, C. T. & Fogel, B. L. Orchestration of neurodevelopmental programs by RBFOX1: implications for autism spectrum disorder. Int Rev Neurobiol. 113, 251–267 (2013).
Cross-Disorder Group of the Psychiatric Genomics et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 45, 984–994 (2013).
Li, J. et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol Psychiatry 21, 290–297 (2015).
Gazzellone, M. J. et al. Copy number variation in Han Chinese individuals with autism spectrum disorder. J Neurodev Disord. 6, 34 (2014).
Chong, W. W. et al. Performance of chromosomal microarray for patients with intellectual disabilities/developmental delay, autism, and multiple congenital anomalies in a Chinese cohort. Mol Cytogenet. 7, 34 (2014).
Girirajan, S. et al. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 7, e1002334, https://doi.org/10.1371/journal.pgen.1002334 (2011).
Yin, C. L. et al. Genome-wide analysis of copy number variations identifies PARK2 as a candidate gene for autism spectrum disorder. Mol Autism 7, 23, https://doi.org/10.1186/s13229-016-0087-7 (2016).
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition. 4th ed edn, (American Psychiatric Association, 1994).
Chien, Y. L. et al. Association study of the CNS patterning genes and autism in Han Chinese in Taiwan. Prog Neuropsychopharmacol Biol Psychiatry. 35, 1512–1517 (2011).
Gau, S. S., Liu, L. T., Wu, Y. Y., Chiu, Y. N. & Tsai, W. C. Psychometric properties of the Chinese version of the social responsiveness scale. Res Autism Spectr Disord. 7, 349–360 (2013).
Chien, W. H. et al. Increased gene expression of FOXP1 in patients with autism spectrum disorders. Mol Autism 4, 23, https://doi.org/10.1186/2040-2392-4-23 (2013).
Chen, C. H. et al. Genetic analysis of GABRB3 as a candidate gene of autism spectrum disorders. Mol Autism. 5, 36, https://doi.org/10.1186/2040-2392-1185-1136. (2014).
Pan, W. H. et al. Han Chinese cell and genome bank in Taiwan: purpose, design and ethical considerations. Hum Hered. 61, 27–30 (2006).
Acknowledgements
We thank the National Center for Genome Medicine of the National Core Facility Program for Biotecnology, Minister of Science and Technology, for the technical support. This work was supported by grants from Ministry of Science and Technology (NSC96-3112-B-002-033, NSC97-3112-B-002-009, NSC98-3112-B-002-004, NSC 99-3112-B-002-036, MOST 103-2314-B-002-055-MY3), National Taiwan University (AIM for Top University Excellent Research Project: 10R81918-03, 101R892103, 102R892103, 103R892103), and National Health Research Institute (NHRI-EX104-10404PI, NHRI-EX105-10404PI), Taiwan.
Author information
Authors and Affiliations
Contributions
Chia-Hsiang Chen and Susan Shur-Fen Gau designed and supervised the study; Hsin-I Chen, and Wei-Hsien Chien conducted the experiments; Ling-Hui Li analyzed the results; Yu-Yu Wu, Yen-Nan Chiu, and Wen-Che Tsai helped with the evaluation and collection of the samples. All authors reviewed drafts and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, CH., Chen, HI., Chien, WH. et al. High resolution analysis of rare copy number variants in patients with autism spectrum disorder from Taiwan. Sci Rep 7, 11919 (2017). https://doi.org/10.1038/s41598-017-12081-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12081-4
This article is cited by
-
Nexus between genome-wide copy number variations and autism spectrum disorder in Northeast Han Chinese population
BMC Psychiatry (2023)
-
LINE-1 and Alu methylation signatures in autism spectrum disorder and their associations with the expression of autism-related genes
Scientific Reports (2022)
-
Identification of copy number variants in children and adolescents with autism spectrum disorder: a study from Turkey
Molecular Biology Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
